08简化测序流程1-有参ddrad
〇.群体遗传学指标
一.准备数据(看4.染色体列表!!!)
1. 测序所在路径,一般有多个文件夹,每个样品一个文件夹
2. 基因组fasta文件,有时需要下载,有时客户提供,还有常见的或者我们做过的可以到 /share/nas6/pub/genome中找找
3. 基因组注释文件,gff3格式
$ perl /share/nas6/xul/program/chloroplast/mvista/genbank.to.gff.pl -i *.gbk -f *.fasta -o tmp.gff #gbk转gff
$ perl -lane '/Name=(.*;)/;print $_."Parent=$1ID=$1"' tmp.gff > genome.gff #修改名字
4. 染色体列表,txt文件,一列
存放序列名称,因为有的fasta文件中有很多序列,该文件用于定义某些图片中需要展示的序列。
保留染色体级别的序列,可以保留!!!所有!!!染色体
如果该基因组都是scaffold,那么就保留几个比较长的序列名称,十个左右就行
$ ir.py -i *.fna -l | sort -k 1 -nr > len.txt
$ cat len.txt | head -n 15 | awk '{print $2}' > chrlist.txt && cat chrlist.txt # 根据情况取前多少行
5. 功能注释结果的整合文件
二.功能注释(有的可以跳过)
可放后台 如果已经做过了,可以跳过该步骤,nas6中的基因组一般都做过这个分析 /share/nas6/pub/genome
2.0 合并步骤
1.挑选cds
gffread -x cds.fa -g *.fna *.gff && perl /share/nas1/yuj/functional_modules/cnvfmt_trans2longest/fasta2longest_with_gff3.pl -i cds.fa -g *.gff -o filter.cds.fa
2.填写配置
cp /share/nas6/pub/pipline/gene-function-annotation/v1.3/profile/fungi_QAll.cfg ./
All_QAll.cfg
BCT_QAll.cfg # 细菌只改绝对路径
danziye_plant_QAll.cfg
fungi_QAll.cfg # 真菌只改绝对路径
Gene_Func_Anno_Pipline.cfg
INV_QAll.cfg
MAM_QAll.cfg
plant_QAll.cfg # 植物改绝对路径 NR/KEGG数据库 双子叶就不用改了
ROD_QAll.cfg
VRL_QAll.cfg # 病毒
VRT_QAll.cfg # 脊椎动物
3.功能注释
nohup perl /share/nas6/pub/pipline/gene-function-annotation/v1.3/Gene_Func_Anno_Pipline.pl --nr --swissprot --cog --kog --kegg --pfam --GO --cfg *_QAll.cfg --od Basic_function &
2.1 获取转录本序列文件
1. 获取转录本序列文件,fasta格式的核酸序列
$ gffread -x cds.fa -g *.fna *.gff
# 遇到错误就删删删 如下
$ grep -n "rna-A564_p001" GCF_004118075.2_ASM411807v2_genomic.gff | wc -l
$ for i in {1..6};do echo $i;sed -i '649514d' GCF_004118075.2_ASM411807v2_genomic.gff;done
# cds要用id来命名,重测序这块分析可能不用管
2.2 获取最长转录本
2.获取最长转录本
有的基因可能存在多个转录本,所以需要过滤下,根据上述提取出来的基因名称判断哪些是同一个基因的,一般id后面有.1 .2这种,就属于一个基因的不同转录本。
具体还要看提取出来的格式。
# perl /share/nas6/zhouxy/functional_modules/cnvfmt_trans2longest/fasta2longest.pl -i cds.fa -o filter.cds.fa # 旧的程序不用 不使用gff,不会改变序列id
$ perl /share/nas6/zhouxy/functional_modules/cnvfmt_trans2longest/fasta2longest_with_gff3.pl -i cds.fa -g *.gff -o filter.cds.fa # 用这个 会把rna的id替换成所属gene的id
$ perl /share/nas1/yuj/functional_modules/cnvfmt_trans2longest/fasta2longest_with_gff3.pl -i cds.fa -g *.gff -o filter.cds.fa # 输出id映射,后面富集分析记得改id
# perl /share/nas6/zhouxy/functional_modules/cnvfmt_trans2longest/fasta_trans_to_geneid_format_convert_ncbi.pl 格式转换 没用过
2.3 填写功能注释的配置文件
3. 填写功能注释的配置文件
# 植物 下面的nr库需要根据情况改成单子叶的或者双子叶的,默认双子叶
$ cp /share/nas6/pub/pipline/gene-function-annotation/v1.3/profile/plant_QAll.cfg ./
# 病毒
$ cp /share/nas6/pub/pipline/gene-function-annotation/v1.3/profile/VRL_QAll.cfg ./
#真菌只改绝对路径
$ cp /share/nas6/pub/pipline/gene-function-annotation/v1.3/profile/fungi_QAll.cfg ./
修改第一行,改成filter.cds.fa的绝对路径!
2.4 运行主程序
4. 运行主程序
$ nohup perl /share/nas6/pub/pipline/gene-function-annotation/v1.3/Gene_Func_Anno_Pipline.pl --nr --swissprot --cog --kog --kegg --pfam --GO --cfg *_QAll.cfg --od Basic_function &
程序运行完后需要用到的文件就是:Basic_function/Result/All_Database_annotation.xls 这个文件,用于填写下面的配置文件
三.主流程
/share/nas6/zhouxy/pipline/genetic_diversity_pip/v1.0/pop_pip_v3.pl
/share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/pop_pip_v3.pl # 循环数 bwa2
3.1 配置文件
示例:
/share/nas6/zhouxy/project/jinhua/GP-20210329-2838_ningdanongxueyuan_malixia_180samples_chicken_ddrad/config.yaml
$ perl /share/nas6/xul/program/ddrad/create_profile_for_ddrad.pl
-i 2.clean_data
-s CM3.6.1_pseudomol.fa
-c chrlist.txt
-gff CM3.6.1_gene.gff3
-a Result/All_Database_annotation.xls
perl /share/nas6/xul/program/ddrad/create_profile_for_ddrad.pl -i 路径/2.clean_data -s data/*.fasta -c data/chrlist.txt -gff data/*.gff -a data/Basic_function/Result/All_Database_annotation.xls
perl /share/nas6/xul/program/ddrad/create_profile_for_ddrad.pl -s data/*.fasta -c data/chrlist.txt -gff data/*.gff -a data/Basic_function/Result/All_Database_annotation.xls -i 路径/2.clean_data
3.2 运行主程序
nohup perl /share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/pop_pip_v3.pl -i ddrad_config.yaml -o analysis -c 1 & # 参考基因组有染色体是-c 1,无染色体时 -c 0
nohup perl /share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/pop_pip_v3_longchr.pl -i ddrad_config.yaml -o analysis -c 1 & # 舍弃了创建HaplotypeCaller结果索引这一步,见4.6章节
3.3 整理结果
perl /share/nas6/zhouxy/pipline/genetic_diversity_pip/current/script/resultdir/resultDir.pl -i analysis/ -o complete_dir
3.4 生成报告
cp /share/nas6/xul/project/ddrad/GP-20210729-3250_ningxiadaxue_211samples_niu_ddrad/pop_html.cfg pop_html.cfg
# 修改配置
perl /share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/html_report_v2/gwas_report2html.pl -id complete_dir/ -cfg pop_html.cfg
# 原版 perl /share/nas6/zhouxy/pipline/genetic_diversity_pip/current/html_report_v2/gwas_report2html.pl -id complete_dir/ -cfg pop_html.cfg
!!!电话要改成18913917953!!!
四.部分结果修改
4.1
cd analysis/variation_dir/variants_stat
Rscript /share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/script/02variant/titv_plot.r --i samples.substitution.xls --o size.samples.substitution
cd analysis/vcf_filter
Rscript /share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/script/05vcf_stat/plot_var_density/popSNPNumStat.r samples.SNPStat.xls_for_plot samples.SNPStat.pdf && convert -density 600 samples.SNPStat.pdf samples.SNPStat.png
cd analysis/vcf_filter
Rscript /share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/script/03vcf/cmplot/cmplot.r --binsize 200000 --input samples.popSNPdensity.list && mv SNP-Density.Col1_Fig1.Col0_Fig1.pdf samples.popSNPdensity.pdf && mv SNP-Density.Col1_Fig1.Col0_Fig1.jpg samples.popSNPdensity.jpg && convert -density 600 samples.popSNPdensity.pdf samples.popSNPdensity.png
cd /share/nas2/yuj/project/2023/ddRAD-seq/GP-20230823-6676_20231023/analysis/PTS_analysis/admixture_dir-qa3
perl /share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/script/10pts/structure/Population_structure.2.0.pl -id /share/nas2/yuj/project/2023/ddRAD-seq/GP-20230823-6676_20231023/analysis/PTS_analysis/admixture_dir-qa3 -o /share/nas2/yuj/project/2023/ddRAD-seq/GP-20230823-6676_20231023/analysis/PTS_analysis/admixture_dir-qa3/samples.admixture.plot.svg
4.2 进化树
perl /share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/script/10pts/tree/vcf2tree.pl -i analysis/vcf_filter/samples.pop.snp.recode.vcf -o analysis/PTS_analysis/tree_dir/samples
FastTreeMP -nt -gtr < analysis/PTS_analysis/tree_dir/samples.fa > analysis/PTS_analysis/tree_dir/samples.phytree.ML.nwk
perl /share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/script/10pts/tree/draw_tree.pl -i /analysis/PTS_analysis/tree_dir/samples.phytree.ML.nwk -o analysis/PTS_analysis/tree_dir/
4.3
四.FAQ
4.1 vcffiles2plink.cmds(20230811 修改之后不会出问题了)
0.生成id映射表
编号目录]
$ bcftools view -H analysis/vcf_filter/samples.pop.snp.recode.vcf | cut -f 1 | uniq | awk '{count++; print $0"\t"count}' | head -n 15 > analysis/PTS_analysis/pca_dir/chrom-map.txt
1.直接生成samples.plink.map
vcftools --vcf analysis/vcf_filter/samples.pop.snp.recode.vcf --plink --chrom-map analysis/PTS_analysis/pca_dir/chrom-map.txt --out analysis/PTS_analysis/pca_dir/samples.plink
2.运行vcffiles2plink.cmds的后半截命令,plink开头的部分
3.
$ touch analysis/.flag_dir/plink2mkbed.ok
4.继续运行主流程
$ perl /share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/pop_pip_v3.pl -i ddrad_config.yaml -o analysis -c 1
4.2 ROH报错 !!!无分组时不进行分析!!! 全都跳过吧
touch analysis/.flag_dir/roh_analysis.ok
尚不知道如何解决,以下步骤无效
1.
bcftools view -H analysis/vcf_filter/samples.pop.snp.recode.vcf | cut -f 1 | uniq | awk '{count++; print $0"\t"count}' > analysis/ROH_dir/chrom-map.txt
chrom-map.txt最后一行不要空行
2.
vcftools --vcf analysis/vcf_filter/samples.pop.snp.recode.vcf --plink --chrom-map analysis/ROH_dir/chrom-map.txt --out analysis/ROH_dir/samples.plink
这一个命令替代ROH.cmds里的第一行命令
修改samples.plink.map,仅保留染色体级别结果
3.接着运行后面3行命令
# xxx /share/nas6/zhouxy/biosoft/plink/v20200428/plink --vcf /share/nas2/yuj/project/2023/ddRAD-seq/GP-20230823-6676_20231023/analysis/vcf_filter/samples.pop.snp.recode.vcf --recode --out /share/nas2/yuj/project/2023/ddRAD-seq/GP-20230823-6676_20231023/analysis/ROH_dir/samples.plink --allow-extra-chr --chr-set 33(染色体条数)
/share/nas6/zhouxy/biosoft/python/3.8.2/bin/python3 /share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/script/14ROH/ped2cnvfmt.py -i analysis/ROH_dir/samples.plink.ped -g analysis/config_dir/group.list -o analysis/ROH_dir/samples.fmt.ped
# /share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/script/14ROH/ped2cnvfmt.py里面最后一个split参数默认是空格,偶尔是制表符
/share/nas6/zhouxy/biosoft/python/3.8.2/bin/python3 /share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/script/14ROH/map2cnvfmt.py -i analysis/ROH_dir/samples.plink.map -o analysis/ROH_dir/samples.fmt.map
/share/nas6/zhouxy/biosoft/R/current/bin/Rscript /share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/script/14ROH/run_dR_cr_ROHom_v2.R /share/nas2/yuj/project/2023/ddRAD-seq/GP-20230823-6676_20231023/analysis/ROH_dir/samples.fmt.ped /share/nas2/yuj/project/2023/ddRAD-seq/GP-20230823-6676_20231023/analysis/ROH_dir/samples.fmt.map 33 /share/nas2/yuj/project/2023/ddRAD-seq/GP-20230823-6676_20231023/analysis/ROH_dir
# 33表示染色体条数
4.3 snpeff出错 不知道错哪就找这一步检查一下
删掉gff中出错的转录本
建库
文件夹结构:
snpEff
├── SnpSift.jar
├── data
│ ├── mp2019
│ │ ├── genes.gff
│ │ └── snpEffectPredictor.bin #建库成功后会显示
│ ├── genomes
│ │ └── mp2019.fa
├── examples/
├── galaxy/
├── scripts/
├── snpEff.config
└── snpEff.jar
java -jar -Xmx50G /share/nas6/zhouxy/biosoft/snpEff/current/snpEff.jar build -c /share/nas1/yuj/project/ddRAD-seq/GP-20220725-4701_20231023/analysis/variation_dir/snpeff_index/sample.config -gff3 -v sample
4.4 sweep分析
补全配置文件,可依据/share/nas1/yuj/project/re-sequencing/GP-20230517-6067_20230621/ref_reseq_config.yaml
Contrast :
- A_vs_B
- A_vs_C
- B_vs_C
4.5 samtools index failed
换成/share/nas1/yuj/software/samtools-0.1.13/samtools这个版本
touch analysis/.flag_dir/samtools_index.ok
perl /share/nas1/yuj/pipline/genetic_diversity_pip/v1.0/pop_pip_v3.pl -i ddrad_config.yaml -o analysis -c 1
4.6 依旧是关于samtools对bam文件建立索引,HaplotypeCaller无法处理过长chr
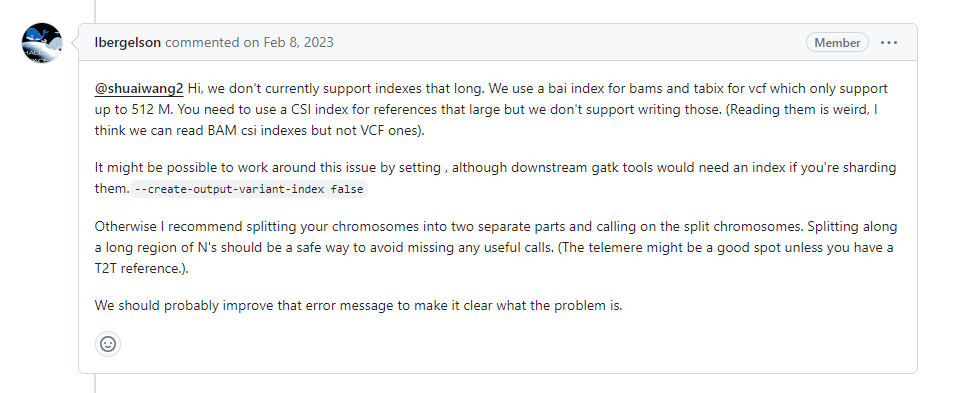
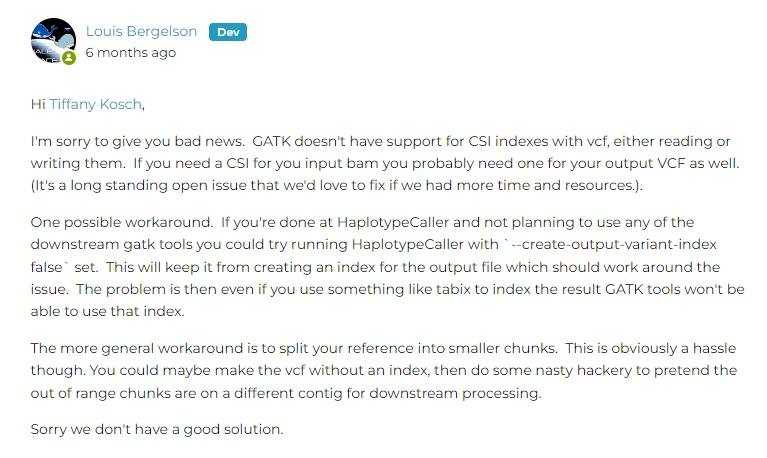
方法1:gatk时不创建索引
/share/nas6/zhouxy/biosoft/GATK/current/gatk --java-options "-Xmx4g" HaplotypeCaller --create-output-variant-index false -R /share/nas2/yuj/project/2024/ddRAD-seq/GP-20240106-7738_20240108/analysis/genome/GWHDUDE00000000.genome.fasta -I /share/nas2/yuj/project/2024/ddRAD-seq/GP-20240106-7738_20240108/analysis/mapping_dir/01.bwa2sortbam/A1.sort.bam -O /share/nas2/yuj/project/2024/ddRAD-seq/GP-20240106-7738_20240108/analysis/variation_dir/HaplotypeCaller/A1.gatk.raw.g.vcf.gz --emit-ref-confidence GVCF -stand-call-conf 30.0 --min-base-quality-score 10 --dont-use-soft-clipped-bases true --tmp-dir
方法2:用bcftools来callsnp(后续怎么做不知道)
bcftools mpileup -Ou -f /share/nas2/yuj/project/2024/ddRAD-seq/GP-20240106-7738_20240108/analysis/genome/GWHDUDE00000000.genome.fasta /share/nas2/yuj/project/2024/ddRAD-seq/GP-20240106-7738_20240108/analysis/mapping_dir/01.bwa2sortbam/A1.sort.bam |bcftools call -mv-o /share/nas2/yuj/project/2024/ddRAD-seq/GP-20240106-7738_20240108/analysis/variation_dir/HaplotypeCaller/0A1.gatk.raw.g.vcf
总结起来
## -----------------5.1 HaplotypeCaller-----------------
1.不要创建HaplotypeCaller结果的索引
--create-output-variant-index false
touch analysis/.flag_dir/gatk_HaplotypeCaller.ok
## -----------------5.2 CombineGVCFs-----------------
2.解压HaplotypeCaller的gz结果
for i in *.gz;do echo $i;gunzip -c $i > ${i%.gz};done
3.对解压后的vcf使用IndexFeatureFile创建idx索引
for i in *.gatk.raw.g.vcf;do gatk IndexFeatureFile -I $i & done
4.CombineGVCFs时加上参数
gatk CombineGVCFs --create-output-variant-index false -R xxx.fna
touch analysis/.flag_dir/gatk_CombineGVCFs.ok
## -----------------5.2 GenotypeGVCFs-----------------
cd /share/nas2/yuj/project/2024/ddRAD-seq/GP-20240106-7738_20240108/analysis/variation_dir/CombineGVCFs
for i in *.gz;do echo $i;gunzip -c $i > ${i%.gz};done
for i in *.g.vcf;do gatk IndexFeatureFile -I $i & done
gatk GenotypeGVCFs --create-output-variant-index false -R xxx
touch analysis/.flag_dir/gatk_GenotypeGVCFs.ok
## -----------------5.3 GatherVCFs-----------------
cd /share/nas2/yuj/project/2024/ddRAD-seq/GP-20240106-7738_20240108/analysis/variation_dir/GenotypeGVCFs
for i in *.gz;do echo $i;gunzip -c $i > ${i%.gz};done
for i in *.raw.vcf;do gatk IndexFeatureFile -I $i & done
sh gatk_GatherVCFs.cmds
touch analysis/.flag_dir/gatk_GatherVCFs.ok
## -----------------5.4 SelectVariants-----------------
包括snp和indel
解压GatherVCFs/samples.gatk.raw.vcf建索引,这里只做snp
-V /share/nas2/yuj/project/2024/ddRAD-seq/GP-20240106-7738_20240108/analysis/variation_dir/GatherVCFs/samples.gatk.raw.vcf ---create-output-variant-index false
touch analysis/.flag_dir/gatk_SelectVariants.ok
## -----------------5.5 VariantRecalibrator-----------------
解压GatherVCFs/samples.gatk.con.snp.vcf.gz然后建索引
第一步,调大java内存,输入文件用解压后的
第二步,输入文件用解压后的,输出不要用压缩格式,如下图所示
java.lang.ArrayIndexOutOfBoundsException: 32772 while running GenotypeGVCFs – GATK
touch analysis/.flag_dir/gatk_VQSR.ok
## Step 5.6 : gatk VariantFiltration
只做snp部分,-V输入-O输出都是未压缩的格式
touch analysis/.flag_dir/gatk_VariantFiltration.ok
## Step 5.7 : gatk Filter
Pipline_sh_commands("gatk_FilterInfo.cmds","gatk_FilterInfo.ok");
只做snp
touch analysis/.flag_dir/gatk_FilterInfo.ok
筛选纯合snp GP-20220805-4753-2杂交种鉴定
1.数据路径
analysis/variation_dir/variants_anno_dir]$
less -S -x20 samples.pop.snp.anno.result.list|grep -v "INTERGENIC"|awk '$5!=$8 && $5!="N" && $8!="N"'|awk '($5=="A" || $5=="T" || $5=="C" || $5=="G") && ($8=="A" || $8=="T" || $8=="C" || $8=="G")'|awk '$6>=10 && $9>=10'|grep SYNONYMOUS_CODING|less -S -x30 > filter.samples.pop.snp.anno.result.list
less -S -x20 samples.pop.snp.anno.result.list # -x20 tab展示为20个空格
|grep -v "INTERGENIC" # 剔除
|awk '$5!=$8 && $5!="N" && $8!="N"' # 不等且不为N
|awk '($5=="A" || $5=="T" || $5=="C" || $5=="G") && ($8=="A" || $8=="T" || $8=="C" || $8=="G")'
|awk '$6>=10 && $9>=10' # 深度大于10
|grep SYNONYMOUS_CODING # 同义编码
|less -S -x30
2.抓取序列
perl /share/nas6/wangyq/src/advance/ssr/get_ssr_region.pl -r region_file(大概是上面filter文件) -g genome -o snp.fasta
3.添加列名
sed -n '1p' samples.pop.snp.anno.result.list > head.txt
4.再加三列Gene_name rawseq snpseq
程序流程
."--------------------- Step 00 : config information -----------------------------\n"
."--------------------- Step 01 : fastQC -----------------------------------------\n"
."--------------------- Step 02 : Reference Index Build --------------------------\n"
."--------------------- Step 03 : BWA Alignment ----------------------------------\n"
."--------------------- Step 04 : BAM statistics --------------------------\n"
."--------------------- Step 5.1 : gatk HaplotypeCaller --------------------------\n"
."--------------------- Step 5.2 : gatk CombineGVCFs and GenotypeGVCFs -----------\n"
."--------------------- Step 5.3 : gatk4 GatherVCFs ------------------------------\n"
."--------------------- Step 5.4 : gatk SelectVariants ---------------------------\n"
."--------------------- Step 5.5 : gatk VariantRecalibrator ----------------------\n"
."--------------------- Step 5.6 : gatk VariantFiltration ------------------------\n"
."--------------------- Step 5.7 : gatk Filter -----------------------------------\n"
."--------------------- Step 6.1 : SnpEff build index ----------------------------\n"
."--------------------- Step 6.2 : GATK Annotation ------------------------------\n"
."--------------------- Step 6.3 : gatk MergeVcfs --------------------------------\n"
."--------------------- Step 6.4 : gatk Result Stat ------------------------------\n"
."--------------------- Step 06 : Vcftools Filter ----------------------------\n"
."--------------------- Step 07 : PTS ---------------------------------------\n"
# 20220916 第一次跑流程,这出了问题
解决:
1.$outdir/vcf_filter/samples.pop.snp.recode.vcf 把没有染色体的删去 该文件是之前步骤生成的
2.再手动执行第7步,造个.ok文件
3.ok 接着跑流程
perl /share/nas6/zhouxy/pipline/genetic_diversity_pip/v1.0/pop_pip_v3.pl -i ddrad_config.yaml -o analysis -c 1
# 下面的命令用来显示map文件里染色体有哪些
a="start" && for i in `awk '{print $2}' /share/nas1/yuj/project/GP-20220715-4649-1_20220825/analysis/PTS_analysis/pca_dir/samples.plink.map | awk -F ':' '{print $1}'`;do if [ $a != $i ];then echo $i ;a=$i;fi;done
NC_040279.1
NC_040280.1
NC_040281.1
NC_040282.1
NC_040283.1
NC_040284.1
NC_040285.1
NC_040286.1
NC_040287.1
NC_040288.1
NC_040289.1
NW_021010809.1
# 下面的命令用来显示map文件里染色体有哪些
n=1;a="NC_040279.1" && for i in `awk '{print $2}' /share/nas1/yuj/project/GP-20220715-4649-1_20220825/analysis/PTS_analysis/pca_dir/samples.plink.map | awk -F ':' '{print $1}'`;do if [ $a != $i ];then a=$i;n=$(expr $n + 1 );elif [ $i = $a ];then echo $i $n;fi;done
NC_040279.1 1
NC_040279.1 1
NC_040279.1 1
...
NC_040289.1 11
NC_040289.1 11
NC_040289.1 11
NC_040289.1 11
NC_040289.1 11
."--------------------- Step 8 : Admixture Structure ------------------------\n"
."--------------------- Step 9 : Phylogenetic tree -------------------------------\n"
."--------- Step 07 : PSMC (Pairwise Sequentially Markovian Coalescent) ---------------\n"
## 整理结果时,这个出问题了 少pdf png
打不开临时文件
然后修改了.bashrc
mkdir /share/nas1/yuj/tmp
export TEMP=/share/nas1/yuj/tmp
."--------------------- Step 07 : Treemix ----------------------------------------\n"
."--------------------- Step 12 : LDdecay ----------------------------------------\n"
."--------------------- Step 13: PopGen statistic --------------------------------\n"
."--------------------- Step 14: ROH Analysis ------------------------------------\n"
## 20220920 第二次跑流程卡住
perl /share/nas6/zhouxy/pipline/genetic_diversity_pip/v1.0/pop_pip_v3.pl -i ddrad_config.yaml -o analysis -c 1
/share/nas6/zhouxy/biosoft/plink/v20200428/plink --vcf $outdir/vcf_filter/samples.pop.snp.recode.vcf --recode --out $outdir/ROH_dir/samples.plink --allow-extra-chr ##这一步之后也是要改map文件的第一列 干脆用上面的map文件来替换 ped文件倒没什么问题
python3 /share/nas6/zhouxy/pipline/genetic_diversity_pip/v1.0/script/14ROH/ped2cnvfmt.py -i $outdir/ROH_dir/samples.plink.ped -g $outdir/config_dir/group.list -o $outdir/ROH_dir/samples.fmt.ped
python3 /share/nas6/zhouxy/pipline/genetic_diversity_pip/v1.0/script/14ROH/map2cnvfmt.py -i $outdir/ROH_dir/samples.plink.map -o $outdir/ROH_dir/samples.fmt.map
/share/nas6/zhouxy/biosoft/R/current/bin/Rscript /share/nas6/zhouxy/pipline/genetic_diversity_pip/v1.0/script/14ROH/run_dR_cr_ROHom_v2.R $outdir/ROH_dir/samples.fmt.ped $outdir/ROH_dir/samples.fmt.map 33 $outdir/ROH_dir ## 流程里是33条染色体,4649-1项目是11条染色体,改成11
造个.ok
ok 接着跑流程
perl /share/nas6/zhouxy/pipline/genetic_diversity_pip/v1.0/pop_pip_v3.pl -i ddrad_config.yaml -o analysis -c 1
."--------------------- Step 6.4 : SnpEff Stat -----------------------------------\n"
## 20220921 第三次跑流程卡住
看那个失败的命令 原因在于软链接的一个文件被挪动了 不要随便改项目的目录结构
把文件挪回去就好了
perl /share/nas6/zhouxy/pipline/genetic_diversity_pip/v1.0/pop_pip_v3.pl -i ddrad_config.yaml -o analysis -c 1
让程序自己生成.ok
这次全部ok了
."--------------------- Step 04 : Variant Merge ----------------------------------\n"
."--------------------- Step 05 : Het Hom ----------------------------------------\n"
."--------------------- Step 06 : Variant TSTV -----------------------------------\n"
."--------------------- Step 06 : Sweep -----------------------------------------\n"
."--------------------- Step 06 : SSR Density ------------------------------------\n"
."--------------------- Step 06 : Population Marker ------------------------------\n"
."--------------------- Step 14: GWAS TASSEL -------------------------------------\n"
."--------------------- Step xxx : MSMC ------------------------------------------\n"
Software
samtools /share/nas6/zhouxy/biosoft/samtools/current/bin/samtools
perl /share/nas6/zhouxy/biosoft/perl/current/bin/perl
python /share/nas6/zhouxy/biosoft/python/2.7.18/bin/python
Rscript /share/nas6/zhouxy/biosoft/R/current/bin/Rscript
ngsqc /share/nas6/zhouxy/biosoft/bin/ngsqc
bwa /share/nas6/zhouxy/biosoft/bwa/current/bwa
bcftools /share/nas6/zhouxy/biosoft/bcftools/current/bin/bcftools
depth_stat_windows /share/nas6/zhouxy/biosoft/bin/depth_stat_windows
gatk /share/nas6/zhouxy/biosoft/GATK/current/gatk
vcftools /share/nas6/zhouxy/biosoft/vcftools/current/bin/vcftools
python3 /share/nas6/zhouxy/biosoft/python/3.8.2/bin/python3
java /share/nas6/zhouxy/software/java/current/bin/java
plink /share/nas6/zhouxy/biosoft/plink/v20200428/plink
PopLDdecay /share/nas6/zhouxy/biosoft/PopLDdecay/current/bin/
primer3_core /share/nas6/zhouxy/biosoft/bin/primer3_core
gcta /share/nas6/zhouxy/biosoft/gcta/current/gcta64
FastTreeMP /share/nas6/zhouxy/biosoft/FastTree/FastTreeMP