05植物线粒体流程2-注释
一.注释
/share/nas6/xul/project/xianliti2/GP-20210812-3293_qinghaidaxue_wangle_2samples_baihe_dasuan_xianliti/re_analysis/assembly/Allium_sativum
构建assembly文件夹,序列名称以$prefix.fasta命名,组装图命名成$prefix.gfa $prefix.gfa.png $prefix.gfa.pdf
!!! 注意.fasta 文件如果是环状需要在id后面增加:circular
prefix=
mkdir -p analysis/{annotation,assembly}/$prefix
cp analysis/assembly/$prefix/$prefix.fasta analysis/annotation/$prefix/
1.1 Gene注释
cd analysis/annotation/$prefix
1.tRNA注释、CDS注释,放后台
有时候xxx/cds/cds_annotation.info无结果,就需要更改线程t的数值
t=8 && nohup perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_mRNA_Gemoma_predict.pl -i *.fasta -o cds -t $t && perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_filter_gemoma_gff_and_2_gene_anno_file.pl -i cds/tmp/final_annotation.gff -o cds/cds_annotation.info & nohup perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_trn_trnascan_predict.pl -i *.fasta -o trna &
2.rRNA注释!!!手动修改好!!!
fl /share/nas6/xul/program/mt2/assembly/database/rrn.fa && blastn -query /share/nas6/xul/program/mt2/assembly/database/rrn.fa -subject *.fasta -outfmt 6 -word_size 25 > rrn.blast && cat rrn.blast && perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_filter_rrn_blast_and_2_gene_anno_file.pl -i rrn.blast > rrn.ann && cat rrn.ann && realpath rrn.ann
然后
blastn -query /share/nas6/xul/program/mt2/assembly/database/rrn.fa -subject *.fasta -outfmt 6 -word_size 25 |cut -f 1,7,8,9,10 > rrna.txt
for j in {rrn5,rrn18,rrn26}; do echo $j; grep -i $j rrna.txt | cut -f 2,3,4,5 | sort -k 3 ;done > allrrna.blast.txt
1 1481 6276 7756
1 1481 6276 7756
从结果里挑选最符合的
3.orf预测(orf不加到gb文件中)
pwd && ssh cluster
cd analyis/annotation/$prefix
/share/nas6/xul/soft/ORFfinder/ORFfinder -in *.fasta -n true -out tmp.orf.fa -outfmt 1 -ml 102 -g 1 && logout
4.合并tRNA,_omit_ann.info,cds/cds_annotation.info
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_merge_muilty_gene_annotation.pl -i trna/*.trna.gene.annotation.info rrn.ann tmp/gene_annotation.info cds/cds_annotation.info > test.ann && readlink -f test.ann
1.2 修改注释
nad1、nad2、nad5三个反式剪接的基因,需要在第五列flag位标注1,其他注释信息依次往后排
其他基因第五列可以直接标其他的flag
PS:
exception="RNA editing" # flag标注为2的含义
note="internal stop codon"
note="stop codon was created by RNA editing"
note="stop codon not determined" / note="start codon not determined"
transl_except="(pos:388686..388688,aa:Met)"
使用第一套密码子
起始密码子:TTG、CTG、ATG(最常见)
终止密码子:TAA、TAG、TGA
atp1 ✔
atp4 ✔
atp6 长度相等,参考ATG组装CTG开头,都是起始,加上transl_except="(pos:complement(92418..92420),aa:Met)"。或者不等往前找ATG
atp8 ✔
atp9 ATG开头;CGA结尾是rna编辑,第五列标2。也可能有TGA结尾的正常拷贝。
ccmB ✔
ccmC、ccmFN、ccmFc
cob
cox1 atg➡️acg flag5=2
cox2、cox3
1977 matR
360 mttB
750 mttB-2
978 nad1
1467 nad2
357 nad3
1488 nad4
303 nad4L
2010 nad5
654 nad6 ✔
1185 nad7
573 nad9
357 sdh3、4
1.挨个基因检查
## 查找某个基因
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_annotation_one_gene_by_ref_gbk2.pl -i2 /share/nas6/xul/project/xianliti2/ref_gbk/Y2.gbk -i1 *.fasta -g xxx基因
## 根据位置提取序列
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_add_gene_seq.pl -i *.fasta -p "3:xxx-yyy:+"
## 这个处理一般也不用
perl -i -F/\\t/ -lane 'BEGIN{sub ee{$nu = shift; if($nu > 436083){ $nu += 1};return $nu}}; $F[1] =~ s/(\d+)/&ee($1)/ge;print join"\t",@F' test.ann # `test.ann` 文件中每一行的第二列中的数字大于436083就+1
## 蛋白比序列* 可以不用,备用的一种方法
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_get_pep_seq_from_gbk.pl -i ../../../../ref2/gbk/*|grep -A 1 rps4 |grep -v "-" > pep.fa
2.根据常见基因,判断基因的丢失
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/find_mt2_miss_gene.pl -i test.ann
3.确定后生成最终的注释文件
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_merge_muilty_gene_annotation.pl -i test.ann -g > gene_annotation.info && realpath gene_annotation.info
1.3 生成gbk
## 生成gbk文件
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_ann2gbk.pl -o gene_anno/ -a gene_annotation.info -f *.fasta -p `basename *.fasta .fasta` && ll gene_anno/
## 多条序列的要合并下,注意合并后的gbk是不能交给客户,只是为了分析方便
perl /share/nas6/xul/program/mt2/annotation/pipline/src/merge_multi_chr_mt_gbk.pl -i gene_anno/{1..40}.gbk > `basename *.fasta .fasta`.gbk
## 单序列的直接复制
cp gene_anno/*.gbk .
二.标准分析
# rm -r tbl rscu_analysis reputer *.gene.annotation.stat.xls *.gc.stat.xls gene_function.xls gene_function2.xls ogdraw
cd analyis/annotation/xxx
下面这些命令全部复制运行
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_get_orf_info_from_ORFfinder_fasta_result.pl -i tmp.orf.fa -a gene_annotation.info -oa orf_annotation.info -of orf.fa && perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_filter_fasta_by_length.pl -i orf.fa -min 300 -o orf_filter.fa && nohup blastx -db /share/nas6/xul/database/nr/Viridiplantae/Viridiplantae.pep.fa -query orf_filter.fa -outfmt 5 -evalue 1e-5 -out blast.pep.xml -num_threads 40 && perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/current/annotation/src/blast_parser.pl -tophit 1 -topmatch 1 -eval 1e-5 -m 7 blast.pep.xml > blast.pep.tophit.xls & \
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_gb2tbl.pl -i gene_anno/`basename *.fasta .fasta`.gbk -o tbl && \
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_extract.feature.pl -i gene_anno/`basename *.fasta .fasta`.gbk -o feature/ -p `basename *.fasta .fasta` && \
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/rscu/rscu_for_cds.pl -i feature/*.cds -o rscu_analysis/ && \
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/repeat/mt2_repeat.pl -mt gene_anno/`basename *.fasta .fasta`.gbk -o reputer/ && \
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_gc_content.pl -i gene_anno/`basename *.fasta .fasta`.gbk -p `basename *.fasta .fasta` > `basename *.fasta .fasta`.gc.stat.xls && \
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_gene_function_with_len.pl -i feature/*.cds feature/*trna feature/*rrna > gene_function.xls && \
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_get_gene_function2.pl -i gene_annotation.info > gene_function2.xls && \
TEMP=./ && nohup perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/ogdraw/ogdraw.pl -i ./gene_anno_for_ogdraw/*.gbk -o ogdraw && \
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_get_cds_seq_use_gene_annotation_file.pl -i gene_annotation.info -f *.fasta -p `basename *.fasta .fasta` -o tmp_for_prep && cd tmp_for_prep && perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/find_mt2_RNAedit_site_like_prep.pl -i *for_perp-mt.txt && \
mv Edited* ../feature && perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_get_edit_site_info.pl -i *_Edit_Sites.txt -p `basename ../*.fasta .fasta` -o ../gene_anno/`basename ../*.fasta .fasta`_final_rna_edit_info.xls && perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_get_RNAediting_type.pl -i ../gene_anno/`basename ../*.fasta .fasta`_final_rna_edit_info.xls > ../gene_anno/`basename ../*.fasta .fasta`_final_rna_edit_info_type.xls && cat ../gene_anno/`basename ../*.fasta .fasta`_final_rna_edit_info.xls |cut -f 2 |grep -v Gene |sort |uniq -c |perl -lane 'BEGIN{print "Gene\tNumber"};print"$F[1]\t$F[0]"' > ../gene_anno/`basename ../*.fasta .fasta`_RNAedit_number.xls && Rscript /share/nas6/xul/program/mt2/annotation/pipline/src/plot_RNAedit_nu.R ../gene_anno/`basename ../*.fasta .fasta`_RNAedit_number.xls ../gene_anno/`basename ../*.fasta .fasta`_RNAedit_number.pdf && convert -density 300 ../gene_anno/`basename ../*.fasta .fasta`_RNAedit_number.pdf ../gene_anno/`basename ../*.fasta .fasta`_RNAedit_number.png &
三.整理结果
3.1 数据质控
cd GP-xxx
touch analysis/samples.reads.txt && readlink -f analysis/samples.reads.txt
填写测序数据路径
samples.reads.txt分为四列:
prefix fq1 fq2 tgs_fq
物种名 r1 r2 3代数据
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/src/mt2_get_result.pl -i analysis -o complete_dir &
## 质控 perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/script/fastq_qc.pl
3.2 生成报告
cp /share/nas6/xul/program/mt2/report/report.cfg ./report.cfg && realpath ./report.cfg
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/report/mt2_report.pl -i complete_dir -cfg ./report.cfg -n # 加上-n表示标准分析
perl /share/nas6/xul/program/mt2/annotation_for_multi_seq/pipline/report/mt2_report_for_hifi.pl -i complete_dir -cfg ./report.cfg -n # 一般不用hifi数据,此命令一般不用
四.补充
4.1 重复序列
4.1.1 散在重复序列柱状图
## 对analysis/annotation/Melicope_pteleifolia/reputer/mt.dispersed.xls操作
grep -v "#" mt.dispersed.xls | sort -k 4 -nr | awk 'BEGIN {OFS="\t"} {print NR, $4, $6, $3, $4, $8, "", $10}' > dispered.repeat.xls
## 生成绘图数据
perl /share/nas6/xul/program/chloroplast/reputer/reputer_state.pl -i dispered.repeat.xls -o dispered.repeat.stat.xls
## 绘图
perl /share/nas1/yuj/program/chloroplast/reputer/draw_reputer_len_and_tye.pl -i *dispered.repeat.stat.xls -o mt.repeat.bar.svg # 去除图例的斜体
svg2xxx -t png -dpi 600 mt.repeat.bar.svg
同目录下存在dispered.repeat.stat.xls文件
python3 /share/nas1/yuj/script/chloroplast/annotation/repeats_ssr_lsr_plot/repeat_plot_bar.py # 直接运行
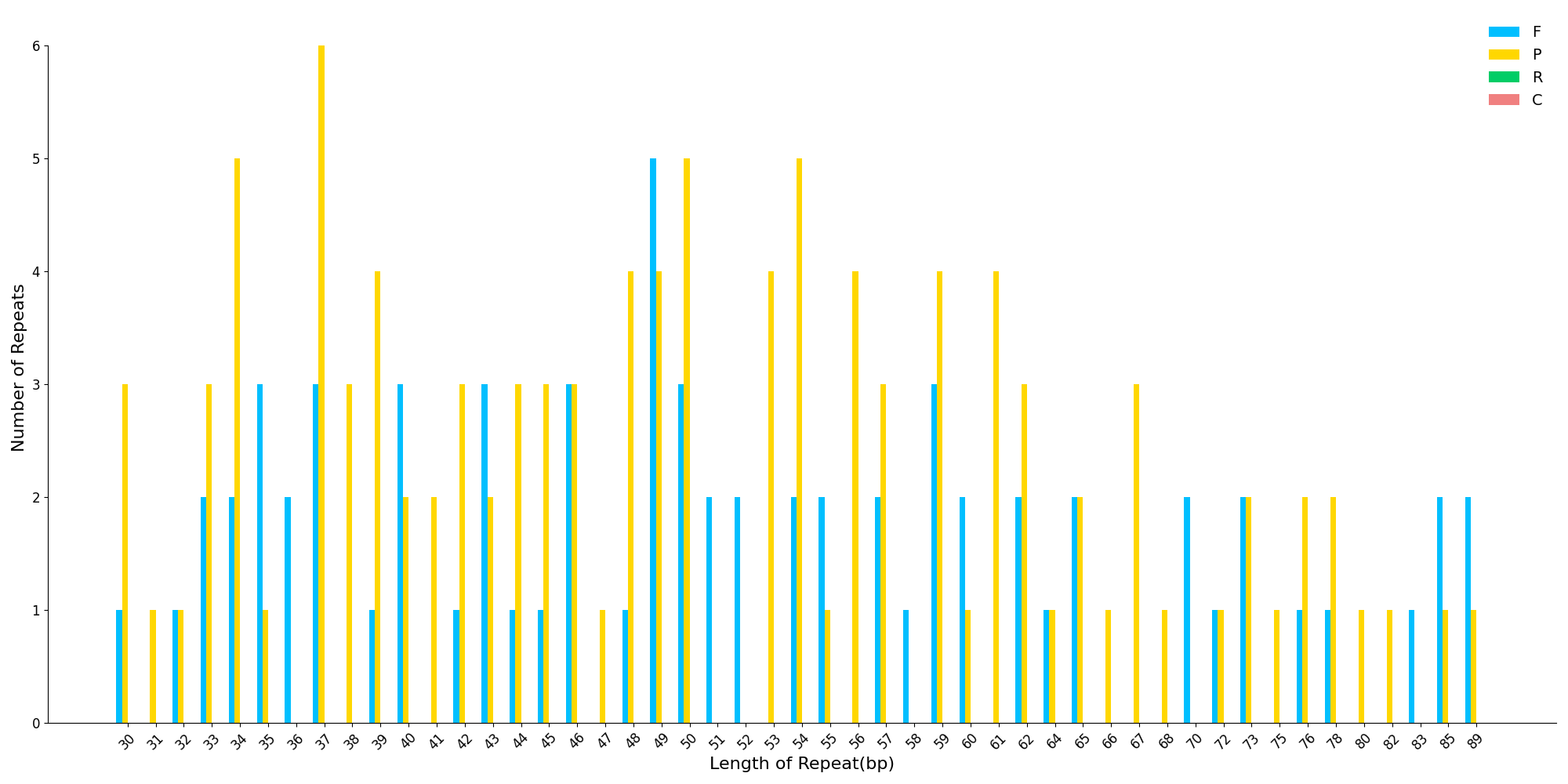
4.1.2 SSR 只针对某个物种绘制
cd analysis/annotation/Melicope_pteleifolia/reputer
Bin="/share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/annotation/bin"
perl $Bin/../etc/misa/src/stat.ssr.pl -i mt.ssr.misa -t *.tbl -o1 ssr.info.xls -o2 ssr.stat.xls
perl $Bin/../etc/misa/src/plot.ssr.stat.noir.pl -i ssr.stat.xls -o ./
perl /share/nas6/xul/program/chloroplast/ssr/draw_ssr.pl -i number.tmp -o ssr.number.bar
cp ssr.stat.xls ./
python3 /share/nas1/yuj/script/chloroplast/annotation/repeats_ssr_lsr_plot/4558_only_ssr_type_stat.py ./ # 获得only_ssr_type_stat.txt
python3 /share/nas1/yuj/script/chloroplast/annotation/repeats_ssr_lsr_plot/ssr_plot.py -i only_ssr_type_stat.txt -o ssr.bar.png
4.2 rna编辑
## 对比原版,去掉边框,数字显示在上方
Rscript /share/nas1/yuj/program/mt2/annotation/pipline/src/plot_RNAedit_nu.R *_RNAedit_number.xls RNAedit_number.pdf && convert -density 300 RNAedit_number.pdf RNAedit_number.png