05线粒体流程2-注释
一.MITOS注释
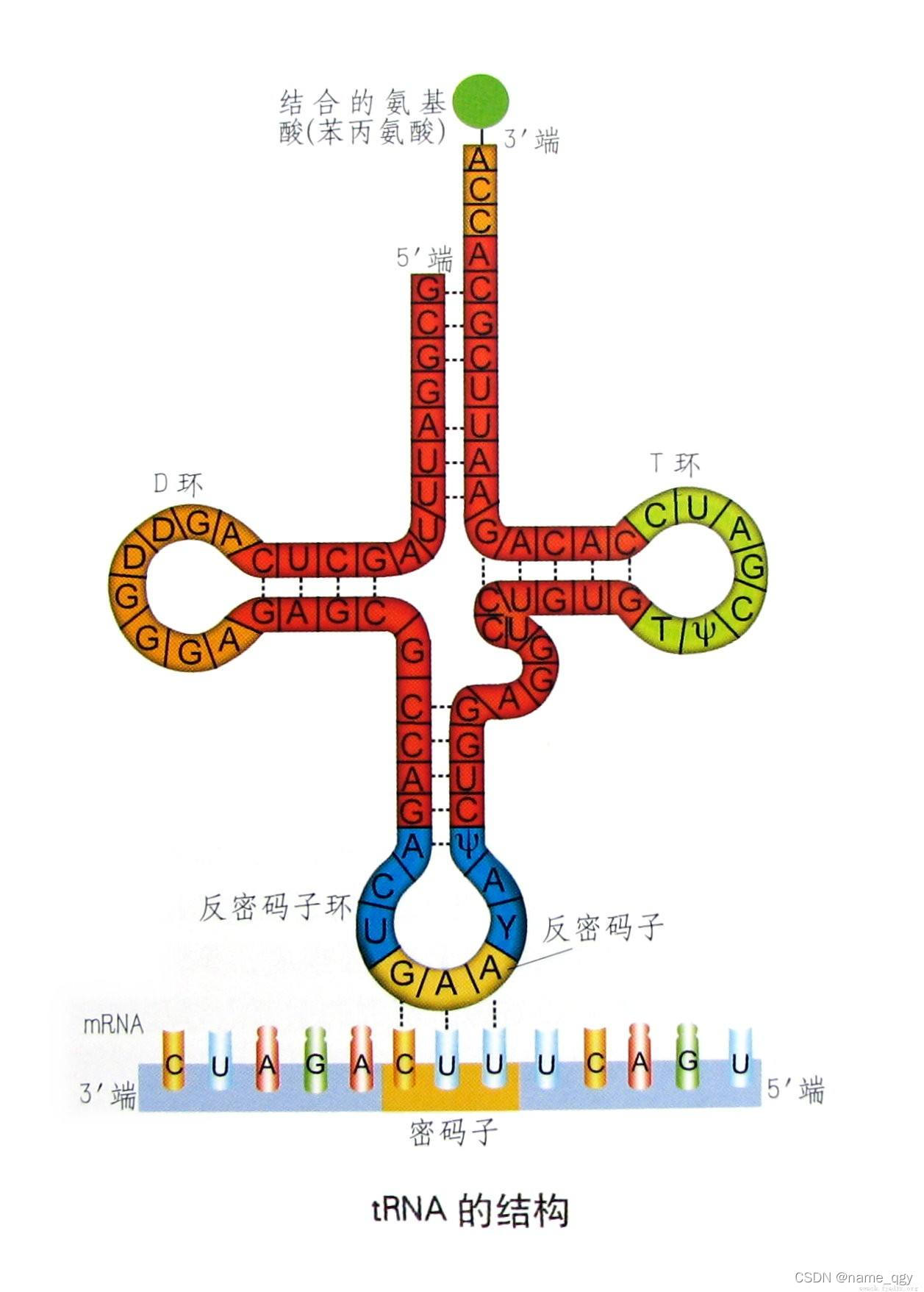
1.0 trna二级结构
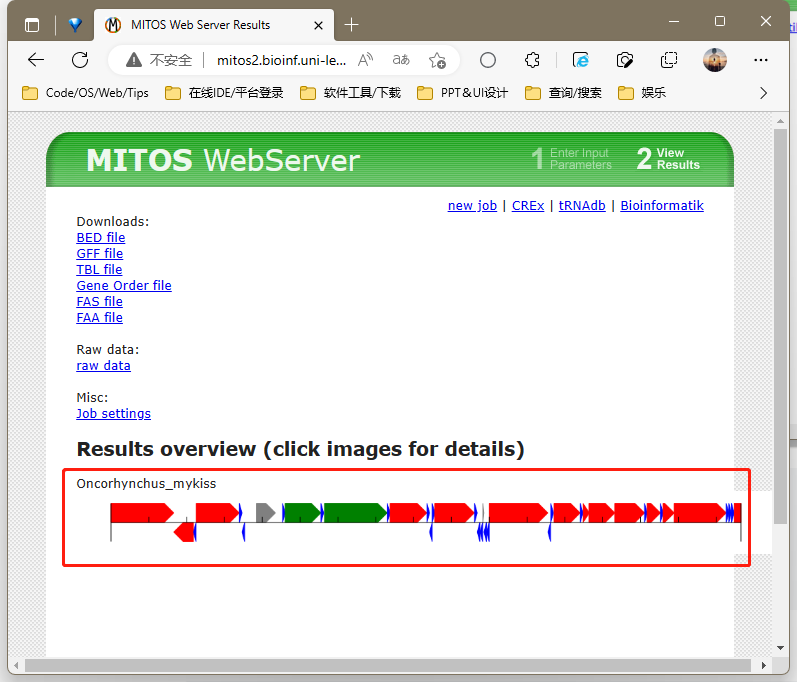
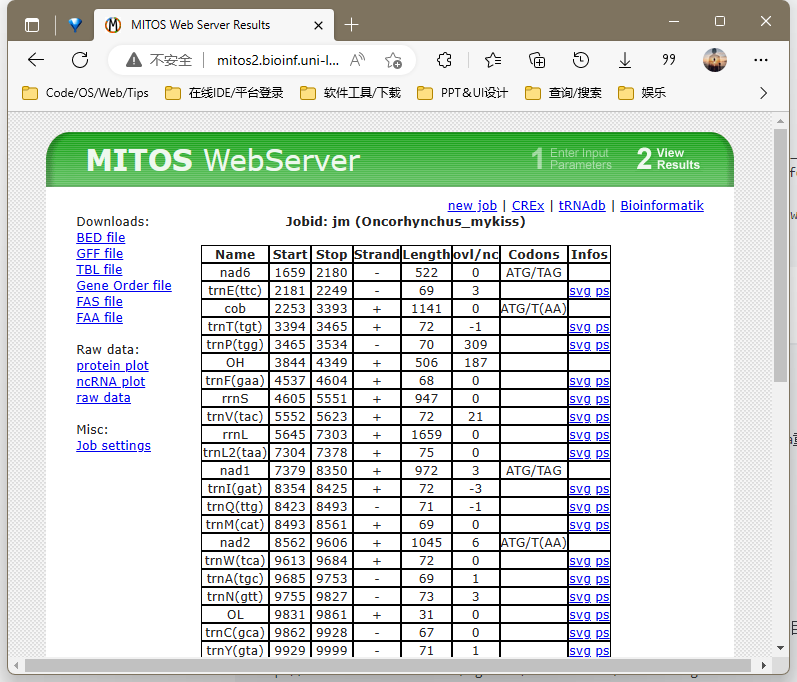
# analysis/annotation/*]$ nohup perl /share/nas6/xul/program/mt/annotation/get_tbl_trnass_from_mitos2.pl -i "网站链接(点击图片后的详情界面)" && cd trna.structure/ # mitos官网已停止服务
1.使用mitos生成的svg来转换
1)上传到galaxy平台,使用mitos注释,下载二级结构
mkdir trna.structure/trn
传到trn目录里
2)重命名
cd trna.structure/trn && rename trn ss-trn *.svg && cd ..
3)修改图片
perl /share/nas6/xul/program/mt/tRNA/draw_tRNA.pl -i trn/*.svg &
2.手动绘制☘️
## 单个绘制
n=s1 && RNAfold < $n.fa > $n.fold
RNAplot -o svg < $n.fold && display *.svg
图片改名 添加trna-反密码子(对该氨基酸所对应密码子的反向互补)
PS:找出的是trna的碱基序列,因此该序列上有的是反密码子,名称就直接写trna-反密码子
## 批量绘制
cd trn
for i in *.fa;do echo ${i%.fa};grep -A 1 ${i%.fa} ../trna_*.fasta > $i ;done
for i in *.fa;do echo ${i%.fa} ; python3 /share/nas2/yuj/project/2024/mitochondrion/GP-20240826-9045-2_20241228/QA1_20250219/trna/a1/trn/3structure.py -i ${i%.fa}.fa -o ${i%.fa}.fold && RNAplot -o svg < ${i%.fa}.fold && rename *_ss.svg ${i%.fa}.svg *_ss.svg; done
3.网页版rnafold
http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi
网页版参数勾选 1 3 3
all_trna_list = ['trnK-TTT', 'trnV-TAC', 'trnL1-TAG', 'trnA-TGC', 'trnP-TGG', 'trnD-GTC', 'trnC-GCA', 'trnF-GAA', 'trnY-GTA', 'trnW-TCA', 'trnG-TCC', 'trnH-GTG', 'trnQ-TTG', 'trnL2-TAA', 'trnN-GTT', 'trnR-TCG', 'trnE-TTC', 'trnM-CAT', 'trnS2-TGA', 'trnS1-GCT', 'trnT-TGT', 'trnI-GAT']
other_trna = {'trnS1':'trnS1-TCT','trnK':'trnK-CTT','trnR':'trnR-ACG'}
# 这一栏填写实际上出现的反密码子,也就是mitos给出的反密码子
1.1 注释
1.for循环步骤
for i in $(ls analysis/annotation);do echo $i;cd analysis/annotation/$i ; mkdir mitos2 ; cd -;done
## 无脊椎
for i in $(ls analysis/annotation);do echo $i;cd analysis/annotation/$i && micromamba run -n mitos2 runmitos.py -i $i"_FULLMT.fsa" -o mitos2/ -c 5 -R /share/nas1/yuj/software/mitos2_refdata/0.3_ref -r refseq63m --maxtrnaovl 100 --maxrrnaovl 100 --rrna 2 & done
## 脊椎
for i in $(ls analysis/annotation);do echo $i;cd analysis/annotation/$i && micromamba run -n mitos2 runmitos.py -i $i"_FULLMT.fsa" -o mitos2/ -c 2 -R /share/nas1/yuj/software/mitos2_refdata/0.3_ref -r refseq63m --maxtrnaovl 100 --maxrrnaovl 100 --rrna 2 & done
2.单个步骤
cd analysis/annotation/xxx
mkdir mitos2
## 无脊椎动物
micromamba run -n mitos2 runmitos.py -i *.fsa -o mitos2/ -c 5 -R /share/nas1/yuj/software/mitos2_refdata/0.3_ref -r refseq63m --maxtrnaovl 100 --maxrrnaovl 100 --best --rrna 2
## 脊椎动物
micromamba run -n mitos2 runmitos.py -i *.fsa -o mitos2/ -c 2 -R /share/nas1/yuj/software/mitos2_refdata/0.3_ref -r refseq63m --maxtrnaovl 100 --maxrrnaovl 100 --rrna 2
二.MITOS注释修改
检查原则(仅供参考)
1.检查所有cds的起止密码子是否正确
末尾是T--,TA-,TAA,都是正确的
有时候末尾不确定的cds,可以往后延伸1bp,也就是“构建出终止子”优先级大于“是否和trna重叠”
举例:
cds 1-100:+ 末尾是ccc
trna 101-170:+ 101上的碱基是T
那么这个cds就是1-101:+,终止子是T--
----------------------------------------------------------------------------------------
2.看重叠,关注cds终点(cds不能与trna重叠)
trna之间可互相重叠
cds之间可相互重叠
若cds与trna方向相同,cds不能与trna重叠,尤其是终点(起点可以落在trna中,但终点不行)
trna 1-71:+
cds 69-200:+或72-200:+
trna 201-271:+
若cds与trna方向不同,终点依旧不能重叠
trna 1-71:-
cds 69-200:+或72-200:+
trna 201-271:-
综上,cds的终点要与trna相接,会出现大量T--,TA-
----------------------------------------------------------------------------------------
3.当缺少trna时,考虑只注释trna,这样能去掉cds干扰
4.删除多余的cds,dloop,确保cds13 trna22 rrna2
5.确定rrna的位置,一般长度和参考一致,一般两端直接与两侧基因首尾相接
6.修改dloop区,大片且连续区域是dloop区,少说也得有个300bp长,长的话10kb也是可能的
要是有两个大片且连续,不确定的话,dloop区可以不标注
dloop要么不标要么标1个
7.画tran二级结构
http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi
RNAfold < test.fa > tmp.fold
RNAplot -o svg < tmp.fold && display *.svg
图片改名
添加trna-反密码子(对该氨基酸所对应密码子的反向互补)
PS:找出的是trna的碱基序列,因此该序列上有的是反密码子,名称就直接写trna-反密码子
网页版参数勾选 1 3 3
8.把生成的trna图片转换后 一并放入文件夹
svg2xxx -t pdf trnH.svg
# xul的一些程序
$ cp_annotation_one_gene_by_ref_gbk2.pl -i1 Thainanus_FULLMT.fsa -i2 ref_ann/gbk/NC_050664.1.gbk -g COX1 # 不看蛋白用叶绿体的找基因或者取序列
$ cp_add_gene_sequence.pl -i ref_ann/fasta/NC_050664.1.fasta -p 74-1039:+ # 只取序列不看蛋白用叶绿体的就行
$ mt_add_gene_sequence.pl -i ref_che/fasta/NC_038207.1.fasta -p 13784-13834 -g ? # 要看蛋白的话,用线粒体,需要指定密码子表
2.0 下载参考
$ vim list_ann
$ down- ann && cd ref_ann/gbk && 2ann- && cd -
# for i in *.gbk ;do echo $i; cp_gbk2ann.pl -i $i -o $i.ann ;done # 抽取参考注释
2.1 解析注释结果
1.for循环步骤
1)转换结果
## 无脊椎
for i in $(ls analysis/annotation);do echo $i;cd analysis/annotation/$i && python3 /share/nas1/yuj/script/mitochondrion/annotation/mt_mitos2info_table.py -i mitos2/result.mitos -fa $i"_FULLMT.fsa" -o gene.info && python3 /share/nas1/yuj/script/mitochondrion/annotation/mt_parse_info_table.py -i gene.info -o gene.annotation.info2 -n 5 && cd - & done
## 脊椎动物
for i in $(ls analysis/annotation);do echo $i;cd analysis/annotation/$i && python3 /share/nas1/yuj/script/mitochondrion/annotation/mt_mitos2info_table.py -i mitos2/result.mitos -fa $i"_FULLMT.fsa" -o gene.info && python3 /share/nas1/yuj/script/mitochondrion/annotation/mt_parse_info_table.py -i gene.info -o gene.annotation.info2 -n 2 && cd - & done
2)看一下基因数量
for i in $(ls analysis/annotation);do echo $i;grep "Total" analysis/annotation/$i/gene.annotation.info2;done
2.单步步骤
1)第一步转换
python3 /share/nas1/yuj/script/mitochondrion/annotation/mt_mitos2info_table.py -i mitos2/result.mitos -fa *.fsa -o gene.info
2)第二步转换
## 无脊椎
python3 /share/nas1/yuj/script/mitochondrion/annotation/mt_parse_info_table.py -i gene.info -o gene.annotation.info2 -n 5
## 脊椎
python3 /share/nas1/yuj/script/mitochondrion/annotation/mt_parse_info_table.py -i gene.info -o gene.annotation.info2 -n 2
2.2 查看基因排列,根据参考修改注释
挑参考
cd gbk
2ann-
grep LOCUS *.gbk
head -n 1 *.ann
2.2.1 共线性好,用一个2023年的参考即可
--- 脊椎动物
count=0 && for i in $(grep 'CDS' gene.annotation.info2 | awk '{print $3 "_:_" $2}'); do count=$((count+1)) && echo "" && echo "###############" $count ":" $(echo "$i" | awk -F "_:_" '{print $1}') "###############" && mt_add.py -i *.fsa -p $(echo "$i" | awk -F "_:_" '{print $2}') -n 2;done > sample_ann.log
count=0 && for i in $(grep 'CDS' *.gbk.ann | awk '{print $3 "_:_" $2}'); do count=$((count+1)) && echo "" && echo "###############" $count ":" $(echo "$i" | awk -F "_:_" '{print $1}') "###############" && mt_add.py -i *.fasta -p $(echo "$i" | awk -F "_:_" '{print $2}') -n 2;done > ref_ann.log
--- 无脊椎动物
count=0 && for i in $(grep 'CDS' gene.annotation.info2 | awk '{print $3 "_:_" $2}'); do count=$((count+1)) && echo "" && echo "###############" $count ":" $(echo "$i" | awk -F "_:_" '{print $1}') "###############" && mt_add.py -i *.fsa -p $(echo "$i" | awk -F "_:_" '{print $2}') -n 5;done > sample_ann.log
count=0 && for i in $(grep 'CDS' *.gbk.ann | awk '{print $3 "_:_" $2}'); do count=$((count+1)) && echo "" && echo "###############" $count ":" $(echo "$i" | awk -F "_:_" '{print $1}') "###############" && mt_add.py -i *.fasta -p $(echo "$i" | awk -F "_:_" '{print $2}') -n 5;done > ref_ann.log
PS:mt_add.py -n 5 -i *.fsa -p # 检查基因,分段的话 -p参数的位置要加双引号
2.2.2 共线性有点差,把所有参考都用上,以无脊椎为例
mkdir ann_tmp
1.提取出cds及对应蛋白序列
python3 /share/nas1/yuj/script/mitochondrion/annotation/mt_from_gbk_get_cds_V2-0.py -i ref_ann/gbk/ -o ref_ann/out -d
cp ref_ann/out/cds/*.fasta ann_tmp/ && python3 /share/nas1/yuj/script/mitochondrion/phytree/mt_extract2mafft_V1.5.py -i ann_tmp/ -o1 gene/extract -o2 gene/mafft -c2
!!!停顿几秒等比对完!!!!
cp *.fsa ann_tmp && cd ann_tmp
for i in cds*.fasta;do ir.py -s7 -n 5 -i $i -o ${i%.fasta}.pep;done # 无脊椎
# for i in cds*.fasta;do ir.py -s7 -n 2 -i $i -o ${i%.fasta}.pep;done # 脊椎
# for i in cds*.fasta;do ir.py -s7 -n 9 -i $i -o ${i%.fasta}.pep;done # 指环虫,9
2.利用核酸和蛋白序列blast比对
for i in cds*.fasta;do blastn -query $i -subject *.fsa -outfmt 6 -max_hsps 1 > $i.log;done
for i in cds*.fasta;do tblastn -query ${i%.fasta}.pep -subject *.fsa -outfmt 6 -max_hsps 1 > ${i%.fasta}.pep.log;done
for i in cds_*.log;do echo $i;cut -f 1,7,8,9,10 $i > ${i%.log}.txt;done
3.进一步处理这些比对出的位置
## for循环直接全部处理了
for gene in {ATP6,ATP8,COX1,COX2,COX3,CYTB,ND1,ND2,ND3,ND4L,ND4,ND5,ND6}; do echo $gene;for i in cds*txt; do grep -i "\b$gene\b" $i ; done | cut -f 2,3,4,5 | sort -k 3 -n ;done > ../allcds.blast.txt && cd ..
1 1481 6276 7756
1 1481 6276 7756
从结果里挑选最符合的
## 比如这个是查找nd4基因
# for i in cds*txt;do grep -i -v nd4l $i| grep -i nd4;done | cut -f 2,3,4,5 | sort -k 3
2.2.3 trna
1.
cp ref_ann/out/trna/*.fasta ann_tmp/ && cd ann_tmp
2.
for i in trna*.fasta;do blastn -task blastn-short -query $i -subject *.fsa -outfmt 6 -max_hsps 1 > $i.log;done
for i in trna_*.log;do echo $i;cut -f 1,7,8,9,10 $i > ${i%.log}.txt;done
3.
## for循环直接全部处理了
for j in {trnL-TAG,trnS-TGA,trnP-TGG,trnT-TGT,trnH-GTG,trnF-GAA,trnE-TTC,trnS-GCT,trnN-GTT,trnR-TCG,trnA-TGC,trnG-TCC,trnD-GTC,trnK-CTT,trnL-TAA,trnY-GTA,trnC-GCA,trnW-TCA,trnQ-TTG,trnI-GAT,trnM-CAT,trnV-TAC}; do echo $j;for i in trna*txt; do grep -i $j $i ; done | cut -f 2,3,4,5 | sort -k 3 -n ;done > ../alltrna.blast.txt && cd .. # 通常不好用
cd ann_tmp
for j in {tRNA-Met,tRNA-Ile,tRNA-Gln,tRNA-Trp,tRNA-Cys,tRNA-Tyr,tRNA-Leu,tRNA-Lys,tRNA-Asp,tRNA-Gly,tRNA-Ala,tRNA-Arg,tRNA-Asn,tRNA-Ser,tRNA-Glu,tRNA-Phe,tRNA-His,tRNA-Thr,tRNA-Pro,tRNA-Ser,tRNA-Leu,tRNA-Val}; do echo $j;for i in trna*txt; do grep -i $j $i ; done | cut -f 2,3,4,5 | sort -k 3 -n ;done > ../alltrna.blast.txt && cd ..
1 1481 6276 7756
1 1481 6276 7756
从结果里挑选最符合的
# tRNA-Ala,tRNA-Arg,tRNA-Asn,tRNA-Asp,tRNA-Cys,tRNA-Gln,tRNA-Glu,tRNA-Gly,tRNA-His,tRNA-Ile,tRNA-Leu,tRNA-Leu,tRNA-Lys,tRNA-Met,tRNA-Phe,tRNA-Pro,tRNA-Ser,tRNA-Ser,tRNA-Thr,tRNA-Trp,tRNA-Tyr,tRNA-Val
tRNA-Ala
tRNA-Arg
tRNA-Asn
tRNA-Asp
tRNA-Cys
tRNA-Gln
tRNA-Glu
tRNA-Gly
tRNA-His
tRNA-Ile
tRNA-Leu
tRNA-Leu
tRNA-Lys
tRNA-Met
tRNA-Phe
tRNA-Pro
tRNA-Ser
tRNA-Ser
tRNA-Thr
tRNA-Trp
tRNA-Tyr
tRNA-Val
## 单独找难找的trna
cd ann_tmp
name="tRNA-Ala"
for i in trna*txt;do grep -i $name $i;done | cut -f 2,3,4,5 | sort -k 3 > $name.log && cat $name.log && for i in trna*fasta;do grep -A 1 -i $name $i;done > $name.fa && fl $name.fa && for i in trna*fasta;do grep -A 1 -i $name $i;done| grep -v ">"
2.2.4 rrna
1.
cp ref_ann/out/rrna/*.fasta ann_tmp && cd ann_tmp
2.
## 通常,相似性高
for i in rrna*.fasta;do blastn -query $i -subject *.fsa -outfmt 6 -max_hsps 1 > $i.log;done
for i in rrna_*.log;do echo $i;cut -f 1,7,8,9,10 $i > ${i%.log}.txt;done
## 一些相似性差的项目
# for i in rrna*.fasta;do blastn -query $i -subject *.fsa -outfmt 6 -max_hsps 1 -task blastn-short > $i.log;done
# for i in rrna_*.log;do echo $i;cut -f 1,7,8,9,10 $i > ${i%.log}.txt;done
3.
for i in rrna*txt;do grep -i 16s $i;done | cut -f 2,3,4,5 | sort -k 3
## for循环直接全部处理了
for j in {large,16S,l-rRNA,rrnL,small,12S,s-rRNA,rrnS}; do echo $j;for i in rrna*txt; do grep -i $j $i ; done | cut -f 2,3,4,5 | sort -k 3 -n ;done > ../allrrna.blast.txt && cd ../
1 1481 6276 7756
1 1481 6276 7756
从结果里挑选最符合的
2.3 没有问题的话,生成gene.annotation.info
## 检查
count=0 && for i in $(grep 'CDS' gene.annotation.info2 | awk '{print $3 "_:_" $2}'); do count=$((count+1)) && echo "" && echo "###############" $count ":" $(echo "$i" | awk -F "_:_" '{print $1}') "###############" && mt_add.py -i *.fsa -p $(echo "$i" | awk -F "_:_" '{print $2}') -n 5;done > sample_ann.log # 无脊椎
## 排序
mt_move_pos.py -i gene.annotation.info2 -o gene.annotation.info
有问题,修改序列起点
/share/nas1/yuj/script/mitochondrion/annotation/mt_move_gene_pos.py
mt_move_pos.py -i gene.annotation.info2 -o gene.annotation.info -n2 49 -m 17121
平移排序注释 帮助信息见-h
PS:以下可以不用管了
trna二级结构有问题,新建一个文件夹进行生成,不要在之前的文件夹里操作,因为会覆盖注释文件!!!
# 如果trna有问题 用以下脚本查找二级结构
$ python3 /share/nas1/yuj/script/mitochondrion/annotation/mt_trnascan_ss_2_rnaflod.py -i *.fsa -o trna.structure/trn # 脚本会输出下一步要做的操作
$ RNAfold < test.fa > tmp.fold
$ RNAplot -o svg < tmp.fold && display *.svg
三.跑注释流程
1.配置文件
20210918]$ python3 /share/nas1/yuj/script/chloroplast/get_ann_cfg.py
# 在项目编号目录下直接运行 或者cp /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/cp.anno.config.yaml ann.cfg
# 修改配置文件,写参考
2.跑流程
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/annotation/mt_annotation_pip.pl -i ann.cfg -g 2 -c
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/annotation/mt_annotation_pip.pl -i ann.cfg -g 5 -c
!!!第一遍流程加-c参数,好用来检查,检查gene.bed.xls
!!!一些特殊情况,如cox1以CGA起始,先修改生成的gbk再接着跑流程
!!!如果确信1.2步骤里做的没问题,可以不加-c,一遍过
/share/nas6/xul/program/mt/bin/mt_get_gene_bed_more.pl -g $gencode -i $indir/annotation/$sample/$sample.gbk > $indir/annotation/$sample/gene.bed.xls
# 程序结果
四.整理结果
1.检查图片
mkdir fig && cp analysis/annotation/*/ogdraw/*.dpi300.circular.png analysis/annotation/*/asmqc/coverage_plot/*.png analysis/annotation/*/asmqc/cmp.dotplot.png fig
2.illumina数据
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/html_report/mt_pip_dir.pl -i analysis
rm -rf complete_dir/03Asmqc/*/*.coverage.svg
2.华大数据
perl /share/nas1/yuj/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/annotation/src/fastq_qc_wgs_bgi.pl -i analysis/samples.reads.txt -o complete_dir/01Rawdata
???原始数据统计 可能出问题???
/share/nas6/zhouxy/modules/ngsqc/ngsqc_bgi -1 fq1 -2 fq2 -k 物种名 -o ./(华大可用.)
/share/nas6/zhouxy/biosoft/bin/ngsqc -1 fq1 -2 fq2 -k 物种名 -o ./ (华大不能用)
Rscript /share/nas6/xul/program/reseq/v1.1/script/ngsqc.R --base *.atgc --qual *.qual --out ./
sub qc{
system "/share/nas6/zhouxy/biosoft/bin/ngsqc -1 $_[1] -2 $_[2] -k $_[0] -o . ";
system "Rscript /share/nas6/xul/program/reseq/v1.1/script/ngsqc.R --base $_[0].atgc --qual $_[0].qual --out $_[0]";
}
for my $svg (@svg){
system "svg2xxx -t pdf $svg.acgtn.svg";
system "svg2xxx -t png -dpi 600 $svg.acgtn.svg";
system "svg2xxx -t pdf $svg.quality.svg";
system "svg2xxx -t png -dpi 600 $svg.quality.svg";
五.生成报告
图片路径 /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/html_report/src/tmp
1.修改报告配置文件
cp /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/html_report/report.cfg ./report.cfg && realpath report.cfg
for i in `cat ann.cfg |grep gbk | cut -f 4`;do echo "#####################"$i && head $i;done
2.标准分析 后面有-n
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/html_report/report2xml.xianliti.pl -id complete_dir/ -cfg report.cfg -n
3.标准分析+高级分析 标准分析用也行
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/html_report/report2xml.xianliti.pl -id complete_dir/ -cfg report.cfg
4.标准分析,不包含测序
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/html_report/report2xml.xianliti2.pl -id complete_dir/ -cfg report.cfg
5.三代辅助组装
## 先单独生成3代统计
perl /share/nas6/xul/program/mt2/tgs_reads_statistic/multi_tgs_qc.pl -i analysis/samples.reads.txt -o complete_dir/01Rawdata/
# samples.reads.txt第四列填3代路径
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/html_report/report2xml.xianliti.3dai.pl -id complete_dir/ -cfg report.cfg
0.容易出问题的质控
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/asmqc/mtDNA_asmqc.pl -i 组装结果 -1 clean*1 -2 clean*2 -p 物种名 -q 物种.gbk -o asmqc -g 2 -r 参考.gbk
# 3490质控出问题,因为忘记改记录原始数据路径的文件了
六.补充
6.1 注释asmqc
## asmqc 深度覆盖圈图 共线性图
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/asmqc/mtDNA_asmqc.pl -i 组装结果 -1 clean*1 -2 clean*2 -p sample -q 物种.gbk -o asmqc -g 2 -r 参考.gbk
## mapping后生成图
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/asmqc/src/pair-end_fastq_mappping.pl -1 /share/nas2/seqloader/xianliti/GP-20241012-9435-4_yunnannongda_1sample_zhu_xianliti/Raw_data/G24/G24_1.fq.gz -2 /share/nas2/seqloader/xianliti/GP-20241012-9435-4_yunnannongda_1sample_zhu_xianliti/Raw_data/G24/G24_2.fq.gz -r analysis/assembly/G24/finish/G24_FULLMT.fsa -s 1 -p G24 -o analysis/annotation/G24/asmqc && perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/asmqc/src/mt2circos.pl -b analysis/annotation/G24/asmqc/mapping/G24.sort.bam -g analysis/annotation/G24/G24.gbk -o analysis/annotation/G24/asmqc/coverage_plot
## 线性覆盖图
cp_bowtie.align.pl -i *.fa -1 *1.fq -2 *2.fq -o ./mapping && cd mapping && samtools view -h -F 4 *.sam > sample.map.sam && samtools sort -@ 8 -o sample.sort.bam -T sample.sss -O bam sample.map.sam && samtools depth sample.sort.bam |awk '{print "mt1",$2,$2,$3}' > sample.depth.txt && python3 /share/nas1/yuj/script/chloroplast/annotation/draw_line_depth_v2.py sample.depth.txt line_depth.png && display line_depth.png && cd - &
6.1.1 测序深度圈图
1.bowtie
cp_bowtie.align.pl -i *.fsa -1 *1.fastq.gz -2 *2.fastq.gz -o analysis/annotation/Lanmaoa_macrocarpa/asmqc/mapping
2.筛选
samtools view -h -F 4 *.sam > sample.map.sam
3.排序
samtools sort -@ 8 -o sample.sort.bam -T sample.sss -O bam sample.map.sam
4.深度统计
samtools depth sample.sort.bam |awk '{print "mt1",$2,$2,$3}' > sample.depth.txt
5.找出深度最大值
awk '{max = ($NF > max) ? $NF : max} END {print max}' sample.depth.txt
6.修改配置文件
cp XXX/J5.circos.conf ./sample.circos.conf
line7修改输出路径
line8修改文件名
112改成新样品信息
207深度绝对路径
210深度最大值
7.画图
/share/nas6/zhouxy/biosoft/circos/current/bin/circos -conf sample.circos.conf
6.1.2 线性深度图
1.asmqc_dir/coverage_plot] $
perl /share/nas6/xul/program/chloroplast/assembly/draw_line_depth.pl *.depth* line.svg && svg2xxx -t png -dpi 600 line.svg && svg2xxx -t pdf line.svg
2.
python3 /share/nas1/yuj/script/chloroplast/annotation/draw_line_depth_v2.py *.depth.txt line_depth.png
6.1.3 共线性图
Bin="/share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/asmqc"
# cmd3 perl $Bin/src/dotplot.pl -r $outdir/refseq.fa -q $infile -o $outdir -p cmp
$ perl $Bin/src/dotplot.pl -r refseq.fa -q $infile -o . -p cmp
# 生成共线性图
# R出错
$ conda env list
$ conda activate R4.2.2
$ perl $Bin/src/dotplot.pl -r refseq.fa -q $infile -o . -p cmp
$ conda deactivate
# cmd4 perl $Bin/src/annotation_mtDNA_genbank.pl -i $infile -r $refgbk -e sample -o $outdir/cmp.gbk
$ perl $Bin/src/annotation_mtDNA_genbank.pl -i $infile -r $refgbk -e sample -o cmp.gbk
# 生成cmp.gbk 和组装的gbk相比里面名称变成了sample
6.2 圈图
6.2.1 OGdraw
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/annotation/etc/plot_ogdraw_cpgenome_genbank/ogdraw.pl -i Ganoderma_sinense.gbk -p Ganoderma_sinense -o ogdraw
真菌项目一些外显子很多的基因,需要改成基因座两端的位置,才能正常画出来
如果有个别基因绘制不出来,是因为/share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/annotation/etc/plot_ogdraw_cpgenome_genbank/conf/OGDraw_chondriom_set.xml配置中没有该基因绘制参数,需要加上
6.2.2 Chloroplot
1.windows分步骤
$ Rscript E:\OneDrive\jshy信息部\Script\chloroplast\R\Win_Chloroplot.R F:\ F:\4939_202301\Ustilago_esculenta_MT10.gbk Ustilago_esculenta_MT10 mt
$ 复制pdf到服务器
$ convert -density 600 *.circular.pdf Ustilago_esculenta_MT10.dpi300.circular.png
2.
使用Chloroplot(https://irscope.shinyapps.io/Chloroplot/)制作线粒体基因组图谱,如下图所示:
注:正向编码的基因位于圈内侧,反向编码的基因位于圈外侧。内部的灰色圈代表GC含量。
3.流程里对这部分的文字描述
#-----4.3.2 线粒体基因组图谱
&EMPTY_TAG('h3','4.3.2 线粒体基因组图谱','','type1');
&EMPTY_TAG('p','使用Chloroplot(https://irscope.shinyapps.io/Chloroplot/)制作线粒体基因组图谱,如下图所示:','type1');
&piclist("图$pid 线粒体基因组图谱",'注:正向编码的基因位于圈内侧,反向编码的基因位于圈外侧。内部的灰色圈代表GC含量。',"$indir/04Annotation/*/ogdraw/*dpi300.circular.png");
6.3 rscu绘图
流程图

cd rscu_analysis
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/annotation/src/rscu.pl -i1 Mm_S3/features/Mm_S3.cds -i2 Mm_S3/features/Mm_S3.pep -g 5 -o Mm_S3/rscu_analysis/rscu.stat.xls
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/annotation/src/plot_rscu_table.pl -i rscu.stat.xls -o rscu.plot.circle.svg -g $gencode
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/annotation/src/plot.rscu.bar.pl -i rscu.stat.xls -o rscu.plot.bar.svg
柱状图+折线
## 加上连线的图
perl /share/nas6/xul/program/chloroplast/rscu/draw.rscu.nu.pl -i rscu.stat.xls -o rscu.svg
svg2xxx -t png -dpi 600 rscu.svg && svg2xxx -t pdf -dpi 600 rscu.svg
流程图更改颜色
## 柱状图
cp /share/nas1/yuj/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/annotation/src/plot.rscu.bar.pl ./
修改颜色
perl plot.rscu.bar.pl -i rscu.stat.xls -o rscu.plot.bar.svg && svg2xxx -t png -dpi 600 rscu.plot.bar.svg && svg2xxx -t pdf -dpi 600 rscu.plot.bar.svg
## 圈图
cp /share/nas1/yuj/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/annotation/src/plot_rscu_table.pl ./
修改颜色
perl plot_rscu_table.pl -i rscu.stat.xls -o rscu.plot.circle.svg -g 2 # 2 或 5
svg2xxx -t png -dpi 600 rscu.plot.circle.svg && svg2xxx -t pdf -dpi 600 rscu.plot.circle.svg
6.4 报告质控
## 测序数据 质控
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/annotation/src/fastq_qc.pl -i analysis/samples.reads.txt -o complete_dir/01Rawdata
6.5 散在重复序列
## 流程
perl /share/nas6/xul/program/mt/repeat/draw_reputer_len_and_tye.pl -i *.dispered.repeat.stat.xls -o repeat.bar.svg && svg2xxx -t pdf repeat.bar.svg && svg2xxx -t png -dpi 300 repeat.bar.svg
## 其他样式
python3 /share/nas1/yuj/script/chloroplast/annotation/repeats_ssr_lsr_plot/plot_repeats_len_and_tye.py -i *.dispered.repeat.stat.xls -o repeat.bar.png
七.批量操作
# for i in $( ls /share/nas1/yuj/project/20211229/GP-20211206-3811/analysis/assembly) ;do echo $i
# 显示site文件的第一列与第二列,以空格分开
for i in `awk -F "\t" '{print $1 $2}' site.txt`;do echo $i;done
# 批量写入shell命令到文件,site.txt里是网址,执行完再单独写到每个.sh文件中
for i in `awk '{print $1}' site.txt`;do echo $i && echo -e "nohup perl /share/nas6/xul/program/mt/annotation/get_tbl_trnass_from_mitos2.pl -i \"$i\" && mt_tbl2ann_mt.pl tmp.tbl >gene.annotation.info && realpath gene.annotation.info &" >> shell.txt;done
# 测试
for i in {g3,j1,j2,j3,l1,l2,l3,s1,s2,s3};do echo $i && cat $i.sh;done
# 批量执行已写好的脚本
for i in {Mm_G3,Mm_J1,Mm_J2,Mm_J3,Mm_L1,Mm_L2,Mm_L3,Mm_S1,Mm_S2,Mm_S3};do echo $i;cd /share/nas1/yuj/project/202201/GP-20211206-3816/analysis/annotation/$i &&pwd && sh /share/nas1/yuj/project/202201/GP-20211206-3816/$i.sh;done
# 批量整理trna二级结构
for i in {Mm_G3,Mm_J1,Mm_J2,Mm_J3,Mm_L1,Mm_L2,Mm_L3,Mm_S1,Mm_S2,Mm_S3};do echo $i;cd /share/nas1/yuj/project/202201/GP-20211206-3816/analysis/annotation/$i/trna.structure/ && pwd && perl /share/nas6/xul/program/mt/tRNA/draw_tRNA.pl -i trn*/*.svg && pwd;done
# 批量blast比对
for i in {Mm_J2,Mm_J3,Mm_L1,Mm_L2,Mm_L3,Mm_S1,Mm_S2,Mm_S3};do echo $i;blastn -query /share/nas1/yuj/project/202201/GP-20211206-3816/r1.fa -subject /share/nas1/yuj/project/202201/GP-20211206-3816/analysis/assembly/$i/finish/*.fsa -outfmt 6;done
# 看组装完的contig深度 和红原鸡线粒体基因组比对 根据深度调整
blastn -query allref.fa -subject *.fsa -outfmt 6
# 批量显示测序数据量
for i in $( ls /share/nas1/yuj/project/20211229/GP-20211206-3811-1_longyanxueyuan_9samples_dongwu_xianliti/analysis/assembly/) ;do echo $i; ll /share/nas1/yuj/project/20211229/GP-20211206-3811-1_longyanxueyuan_9samples_dongwu_xianliti/analysis/assembly/$i/pseudo/$i/1_Trimmed_Reads/$i.trimmed_P1.fq && ll /share/nas1/yuj/project/20211229/GP-20211206-3811-1_longyanxueyuan_9samples_dongwu_xianliti/analysis/assembly/$i/pseudo/$i/1_Trimmed_Reads/$i.trimmed_P2.fq ;done
# 批量显示比对结果
for i in $( ls /share/nas1/yuj/project/20211229/GP-20211206-3811-1_longyanxueyuan_9samples_dongwu_xianliti/analysis/assembly/) ;do echo $i; blastn -query /share/nas1/yuj/project/20211229/GP-20211206-3811-1_longyanxueyuan_9samples_dongwu_xianliti/ref_ass/all_ref.fa -subject /share/nas1/yuj/project/20211229/GP-20211206-3811-1_longyanxueyuan_9samples_dongwu_xianliti/analysis/assembly/$i/pseudo/$i/1_Trimmed_Reads/uni/assembly.fasta -outfmt 6 ;done
# 批量质控
for i in $( ls analysis/assembly/) ;do echo $i; perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/asmqc/mtDNA_asmqc.pl -i ref_ass/fasta/HQ857211.1.fasta -1 /share/nas1/seqloader/xianliti/GP-20211206-3811-1_longyanxueyuan_9samples_dongwu_xianliti/2.clean_data/$i/$i_*_R1_001.fastq.gz -2 /share/nas1/seqloader/xianliti/GP-20211206-3811-1_longyanxueyuan_9samples_dongwu_xianliti/2.clean_data/$i/$i_*_R2_001.fastq.gz -p Gallus_gallus -q /share/nas1/yuj/project/20211229/GP-20211206-3811-1_longyanxueyuan_9samples_dongwu_xianliti/ref_ass/gbk/HQ857211.1.gbk -o /share/nas1/yuj/project/20211229/GP-20211206-3811-1_longyanxueyuan_9samples_dongwu_xianliti/analysis/annotation/$i/asmqc -g 2 -r /share/nas1/yuj/project/20211229/GP-20211206-3811-1_longyanxueyuan_9samples_dongwu_xianliti/ref_ass/gbk/MT800385.1.gbk;done
1.MITOS官方停止服务
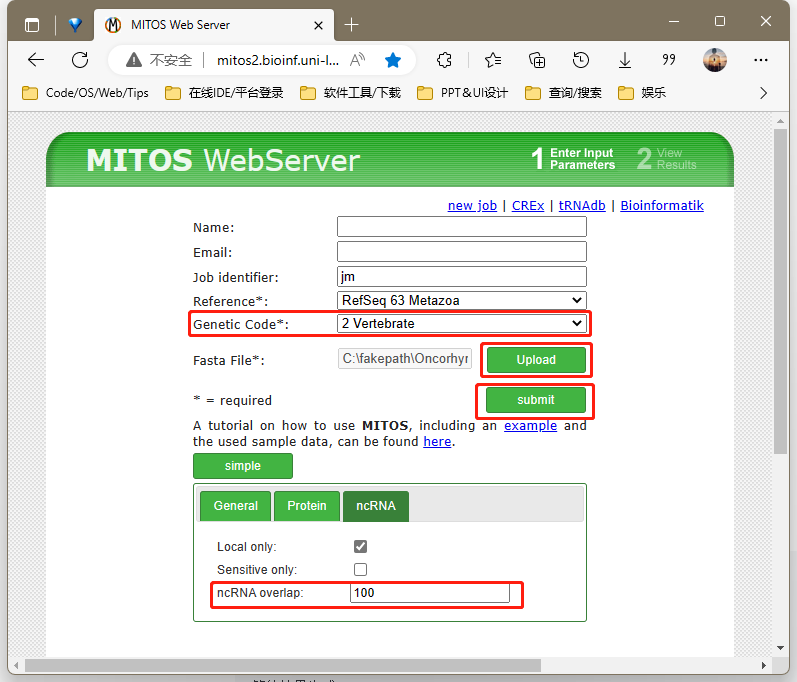
打开http://mitos2.bioinf.uni-leipzig.de/index.py
Reference 选择63
Genetic Code 选择5无脊椎动物or脊椎动物2
点击upload 上传fasta文件
高级选项ncRNA overlap 改100
点击submit 等待结果生成