00VCF文件
一. VCF文件
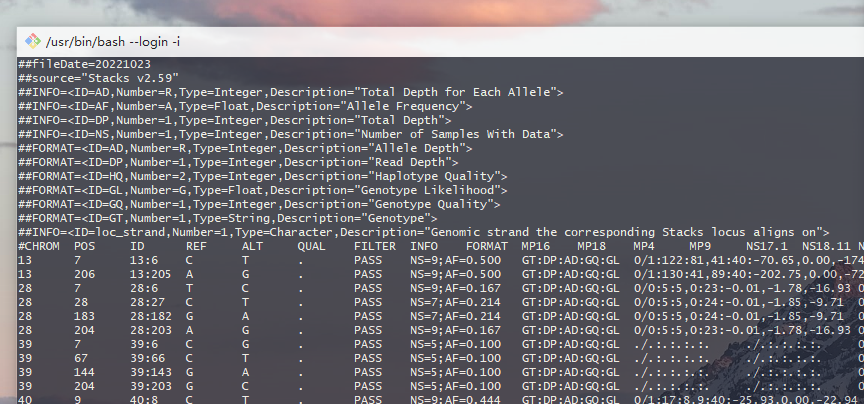
1.1 简介
以“#”开头的注释部分:VCF的介绍信息,通常以##作为起始,其后一般接以FILTER,INFO,FORMAT等字样。
以##FILTER开头的行,表示注释VCF文件当中第7列中缩写词的说明,比如q10为Quality below 10;##INFO开头的行注释VCF第8列中的缩写字母说明,比如AF代表Allele Frequency也就是等位基因频率;##FILTER开头的行注释VCF第9列中的缩写字母说明;另外还有其他的一些信息,文件版本"fileformat=VCFv4.0"等等。
没有“#”开头的主体部分:每一行代表一个variant,各列之间用tab空白隔开,前面9列为固定列,第10列开始为样品信息列,可以无限多个
# 主体部分10列的范例
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT MP16
13 7 13:6 C T . PASS NS=9;AF=0.500 GT:DP:AD:GQ:GL 0/1:122:81,41:40:-70.65,0.00,-174.69
1.2 10列分别代表的意义
1.CHROM:染色体编号
2.POS:variant所在的left-most位置(1-base position,发生变异的位置的第一个碱基所在的位置)
3.ID:variant的ID,对应着dbSNP数据库中的ID。(SNP/INDEL的dbSNP编号通常以rs开头,一般只有人类基因组才有dbSNP编号,若没有,则默认使用‘.’)
4.REF:参考序列的Allele。(等位碱基,即参考序列该位置的碱基类型及碱基数量,必须是A,C,G,T,N且都大写)
5.ALT:variant的Allele,若有多个,则使用逗号分隔。(变异的碱基类型及碱基数量)这里的碱基类型和碱基数量,对于SNP来说是单个碱基类型的编号,而对于Indel来说是指碱基个数的添加或缺失,以及碱基类型的变化
6.QUAL:variants的质量。PPhred格式(Phred_scaled)的质量值,代表着此位点是纯合的概率,此值越大,则纯合概率越低,表示突变可能性越大。(表示变异碱基的可能性)
计算方法:Phred值 = -10 * log (1-p), p为variant存在的概率; 通过计算公式可以看出值为10的表示错误概率为0.1,该位点为variant的概率为90%。
7.FILTER:使用上一个QUAL值来进行过滤的话,是不够的。GATK能使用其它的方法来进行过滤,过滤结果中通过则该值为”PASS”;若variant不可靠,则该项不为”PASS”或为”.”。
8.INFO:variant的相关信息
9.FORMAT:variants的格式,例如GT:AD:DP:GQ:PL
10.SAMPLES:各个Sample的值,由BAM文件中的@RG下的SM标签所决定,这些值对应着第9列的各个格式,不同格式的值用冒号分开,每一个sample对应着1列;多个samples则对应着多列,这种情况下列的数多余10列。
1.3 FORMAT 及对应 SAMPLES 值说明
FORMAT字段与样本数据列共同构成基因型信息,格式为 FORMAT字段:样本数据。例如:
GT:AD:DP:GQ:PL 0/1:6,5:11:99:138,0,153
1. GT(GenoType)
格式特征:
- 使用
|或/分隔两个数字(二倍体) - 单倍体(如叶绿体基因组)直接显示单一数字
|表示 Phased genotype(明确等位基因来源的染色体)/表示 Unphased genotype(未明确染色体来源)
数值含义:
| 基因型 | 二倍体含义 | 单倍体含义 |
|---|---|---|
0 |
- | 与REF一致 |
1 |
- | 与ALT一致 |
0/0 |
纯合REF | 不适用 |
0/1 |
杂合REF/ALT | 不适用 |
1/1 |
纯合ALT | 不适用 |
./. |
数据缺失 | 数据缺失 |
2. AD(Allele Depth)
数据格式:逗号分隔的数值
含义:各等位基因的reads覆盖深度
二倍体示例:6,5 → REF深度6,ALT深度5
3. DP(Depth)
计算方式:DP = SUM(AD)
单倍体特殊性:可能直接表示唯一等位基因深度
4. GQ(Genotype Quality)
特性:
- Phred格式质量值(
GQ = -10 * log10(1-P),P为基因型正确概率) - 单倍体与二倍体计算规则相同
5. PL(Genotype Likelihoods)
数据格式:
- 二倍体:逗号分隔的三个值(对应
0/0, 0/1, 1/1) - 单倍体:逗号分隔的两个值(对应
0, 1)
计算规则:PL = -10 * log10(p)(p为对应基因型概率)
1.4 INFO 信息列
AC=1;AF=0.500;AN=2;BaseQRankSum=0.748;ClippingRankSum=0.000;DB;DP=34;ExcessHet=3.0103;FS=3.424;MLEAC=1;MLEAF=0.500;MQ=31.07;MQRankSum=-0.087;QD=11.87;ReadPosRankSum=-1.349;SOR=2.636
AC=2;AF=1.00;AN=2;DB;DP=14;ExcessHet=3.0103;FS=0.000;MLEAC=2;MLEAF=1.00;MQ=31.60;QD=29.36;SOR=5.421
| TAG | 描述 | 示例值 | 备注 |
|---|---|---|---|
| AC | 等位基因计数(Allele Count),与变异一致的Allele总数 | AC=1, AC=2 | 双倍体样本中,0/1基因型对应AC=1,1/1对应AC=2 |
| AF | 等位基因频率(Allele Frequency),AF = AC / AN | AF=0.500, AF=1.00 | 双倍体样本中,0/1基因型对应AF=0.5,1/1对应AF=1.0 |
| AN | 总等位基因数(Allele Number) | AN=2 | 双倍体样本固定为2 |
| BaseQRankSum | Alt与Ref碱基质量的Wilcoxon秩和检验Z值 | 0.748 | 负值表示Alt碱基质量低于Ref |
| ClippingRankSum | Alt与Ref硬剪切碱基数的Wilcoxon秩和检验Z值 | 0.000 | 用于检测剪切偏好性 |
| DB | 标记该变异位点存在于公共数据库中 | 无具体值 | 仅存在标记(如"DB")表示True |
| DP | 过滤后reads覆盖深度 | DP=34, DP=14 | 区别于未过滤的原始DP |
| ExcessHet | 过量杂合性的Phred标准化p值 | 3.0103 | 值越大越可能错误 |
| FS | Fisher精确检验的链偏好性Phred值 | 3.424, 0.000 | 值越大链偏好性越显著,推荐保留FS < 10-20 |
| MLEAC | 最大似然估计的等位基因计数 | MLEAC=1, MLEAC=2 | 可能与AC不同 |
| MLEAF | 最大似然估计的等位基因频率 | MLEAF=0.500 | 可能与AF不同 |
| MQ | 比对质量的均方根值(RMS Mapping Quality) | 31.07, 31.60 | 值越高比对质量越好 |
| MQRankSum | Alt与Ref比对质量的Wilcoxon秩和检验Z值 | -0.087 | 负值表示Alt比对质量差于Ref,推荐保留 > -1.65~-3.0 |
| QD | 深度标准化变异质量(Variant Confidence/Quality by Depth) | 11.87, 29.36 | 值越高可信度越高 |
| ReadPosRankSum | Alt与Ref读段位置的Wilcoxon秩和检验Z值 | -1.349 | 负值表示Alt更靠近reads末端,推荐保留 > -1.65~-3.0 |
| SOR | 链偏倚对称比值比(Symmetric Odds Ratio) | 2.636, 5.421 | 值越大链偏好性越强(>3表示可能存在偏好性) |
| Dels | 含跨缺失reads的比例 | 未显示示例 | 值为0表示SNP,无此TAG表示INDEL |
| HaplotypeScore | 与最多两个分离单倍型的一致性 | 未显示示例 | 值越高表示单倍型不一致风险越高 |
| InbreedingCoeff | 基于哈迪-温伯格平衡的近交系数 | 未显示示例 | 用于检测群体结构异常 |
| MQ0 | 比对质量为零的reads总数 | 未显示示例 | 值高可能表示比对问题 |
| RPA | 串联重复单元的重复次数(每个等位基因) | 未显示示例 | 与STR相关 |
| RU | 串联重复单元序列 | 未显示示例 | 例如"RU=GAA"表示重复单元为GAA |
| STR | 标记短串联重复变异 | 未显示示例 | 存在标记表示该变异为短串联重复 |
关键过滤建议:
- FS:推荐保留 < 10-20
- MQRankSum:推荐保留 > -1.65 ~ -3.0
- ReadPosRankSum:推荐保留 > -1.65 ~ -3.0
- QD:通常过滤低QD值(如 < 2)
- SOR:> 3时可能存在链偏好性风险