samtools
## bam转换为fq
samtools fastq -1 mapped_1.fq -2 mapped_2.fq -s mapped_singles.fq ids1-1.Chr3-34258193-34462334.sort.bam
## sam排序并直接输出为bam(sort功能默认输出为bam,-O参数默认bam)
samtools sort $i".sam" > $i".bam"
## 提取scaffold1上能比对到30k到100k区域的比对结果 [samtools软件从bam文件提取目标序列 - 简书](https://www.jianshu.com/p/c97df2e16642)
samtools view -b -h DJ.sort.bam Chr3:34258193-34462334 > dj.Chr3-34258193-34462334.sort.bam
# 去掉“-b”选项则输出为sam
## 提取基因组指定区间
samtools faidx *.fna
samtools faidx *.fna chr1:1000000-2000000 > chr1_1000000_2000000.fasta
## 根据样品拆分vcf
bgzip Evo.recode.vcf
tabix -p vcf Evo.recode.vcf.gz
bcftools view -S DB_sample.list samples.gatk.con.snp.12.vcf -O v -o samples.gatk.con.DB.snp.vcf
for i in *.txt;do echo ${i%.txt};bcftools view -S $i Evo.recode.vcf.gz -O v -o ${i%.txt}.vcf & done
安装
$ wget https://github.com/samtools/samtools/releases/download/1.9/samtools-1.9.tar.bz2
$ tar xvfj samtools-1.9.tar.bz2
$ cd samtools-1.9
$ ./configure --prefix=/where/to/install
$ make
$ make install
1. 查看(viewing)
1.1 view(用于提取/转换)
1.1.1 参数
Usage: samtools view [options] <in.bam>|<in.sam>|<in.cram> [region ...]
Options:
-b output BAM
-C output CRAM (requires -T)
-1 use fast BAM compression (implies -b)
-u uncompressed BAM output (implies -b)
-h include header in SAM output
-H print SAM header only (no alignments)
-c print only the count of matching records
-o FILE output file name [stdout]
-U FILE output reads not selected by filters to FILE [null]
-t FILE FILE listing reference names and lengths (see long help) [null]
-L FILE only include reads overlapping this BED FILE [null]
-r STR only include reads in read group STR [null]
-R FILE only include reads with read group listed in FILE [null]
-q INT only include reads with mapping quality >= INT [0]
-l STR only include reads in library STR [null]
-m INT only include reads with number of CIGAR operations consuming
query sequence >= INT [0]
-f INT only include reads with all of the FLAGs in INT present [0]
-F INT only include reads with none of the FLAGS in INT present [0]
-G INT only EXCLUDE reads with all of the FLAGs in INT present [0]
-s FLOAT subsample reads (given INT.FRAC option value, 0.FRAC is the
fraction of templates/read pairs to keep; INT part sets seed)
-M use the multi-region iterator (increases the speed, removes
duplicates and outputs the reads as they are ordered in the file)
-x STR read tag to strip (repeatable) [null]
-B collapse the backward CIGAR operation
-? print long help, including note about region specification
-S ignored (input format is auto-detected)
--input-fmt-option OPT[=VAL]
Specify a single input file format option in the form
of OPTION or OPTION=VALUE
-O, --output-fmt FORMAT[,OPT[=VAL]]...
Specify output format (SAM, BAM, CRAM)
--output-fmt-option OPT[=VAL]
Specify a single output file format option in the form
of OPTION or OPTION=VALUE
-T, --reference FILE
Reference sequence FASTA FILE [null]
-@, --threads INT
Number of additional threads to use [0]
1.1.2 提取
# 提取
samtools view -F 4 extend.sam | perl -lane 'print unless($F[9] eq "*")' | perl -ane 'print if(/^@/);if(/NM:i:(\d+)/){$n=$1;$l=0;$l+=$1 while $F[5]=~ /(\d+)[M]/g;if($l > 1000){print}}'|sort -k 4 -n> tmp.sam && cat tmp.sam |cut -f 10 |perl -lane 'print ">",++$i;print $F[0]' > map_gene.fa
# $l > 1000 长度大于1K
-F 数字4代表该序列比对到参考序列上 数字8代表该序列的mate序列比对到参考序列上 ???
# 提取比对到参考序列上的比对结果
$ samtools view -bF 4 abc.bam > abc.F.bam
# 提取paired reads中两条reads都比对到参考序列上的比对结果,只需要把两个4+8的值12作为过滤参数即可
$ samtools view -bF 12 abc.bam > abc.F12.bam
# 提取没有比对到参考序列上的比对结果
$ samtools view -bf 4 abc.bam > unmapped.bam
# 比对到反向互补链的reads
$ samtools view -f 16 test.bam|head -1
# 比对到正向链的reads
$ samtools view -F 16 test.bam|head -1
# 统计共有多少条reads(pair-end-reads这里算一条)参与了比对参考基因组
$ samtools view -c test.bam
# 筛选出比对失败的reads,看序列特征
$ samtools view -f 4 test.bam|cut -f10 |head -3
# 筛选出比对质量值大于30的情况
$ samtools view -q 30 test.bam |awk '{print $1,$5}'|head -3
# 筛选出比对成功,但是并不是完全匹配的序列
$ samtools view -F 4 test.bam |awk '{print $6}'|grep '[IDNSHPX]'|head -5
1.1.3 转换
# sam转bam
samtools view -Sb xxx.sam > xxx.bam
samtools view -b -S xxx.sam -o xxx.bam
# bam转sam
samtools view -h xxx.bam > xxx.sam
# 提取bam文件中比对到caffold1上的比对结果,并保存到sam文件格式
samtools view abc.bam scaffold1 > scaffold1.sam
# 提取scaffold1上能比对到30k到100k区域的比对结果
samtools view abc.bam scaffold1:30000-100000 > scaffold1_30k-100k.sam
# 根据fasta文件,将 header 加入到 sam 或 bam 文件中
samtools view -T genome.fasta -h scaffold1.sam > scaffold1.h.sam
1.2 tview(交互式查看)
交互式的查看reads比对到参考基因组上的信息,类似IGV
1.2.1 参数
Usage: samtools tview [options] <aln.bam> [ref.fasta]
Options:
-d display output as (H)tml or (C)urses or (T)ext
-p chr:pos go directly to this position
-s STR display only reads from this sample or group
--input-fmt-option OPT[=VAL]
Specify a single input file format option in the form
of OPTION or OPTION=VALUE
--reference FILE
Reference sequence FASTA FILE [null]
1.2.2 举例
samtools tview SRR2584866.aligned.sorted.bam ecoli_ref.fa
#result:
1 11 21 31 41 51 61 71 81 91 101 111 121
AGCTTTTCATTCTGACTGCAACGGGCAATATGTCTCTGTGTGGATTAAAAAAAGAGTGTCTGATAGCAGCTTCTGAACTGGTTACCTGCCGTGAGTAAATTAAAATTTTATTGACTTAGGTCACTAAATAC
..................................................................................................................................
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ..................N................. ,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,........................
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ..................N................. ,,,,,,,,,,,,,,,,,,,,,,,,,,,.............................
...................................,g,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, .................................... ................
结果:第一行是ref的位置号,第二行是ref序列(如果不在命令中添加ref的fa文件,这一行将显示N),之后是比对情况,‘.’表示正链匹配到参考基因组上,逗号‘,’表示反链匹配到参考基因组上,‘.’中间出现大写的ATCGN代表该碱基没有匹配上,‘,’中间出现小写的atcgn代表该碱基没有匹配上。按ctrl+c或q退出。
2.索引(indexing)
在NGS数据分析过程中,包括比对在内的很多地方都需要建索引,主要作用就是为了快,提高效率。对一些大文件我们要养成建索引的习惯,况且一些功能的正常运行必需要提供索引文件,有的软件在生成结果文件的同时也生成其对应的索引文件,可以说,索引随处可见。
2.1 faidx(针对.fasta)
对fasta参考基因组建索引(faidx)
$ samtools faidx ref.fa
# 生成ref.fa.fai
2.2 index(针对.BAM)
2.2.1 参数
Usage: samtools index [-bc] [-m INT] <in.bam> [out.index]
Options:
-b Generate BAI-format index for BAM files [default]
-c Generate CSI-format index for BAM files
-m INT Set minimum interval size for CSI indices to 2^INT [14]
-@ INT Sets the number of threads [none]
2.2.2 举例
对BAM文件建索引
$ samtools index test.sorted.dup.bam
# 生成test.sorted.dup.bam.bai
3. 文件操作(file operations)
3.1 sort(排序)
当利用FASTQ文件与参考基因组进行比对时,reads比对结果的顺序相对于它们在参考基因组中的位置而言是随机的,也就是说BAM文件按照序列在输入的FASTQ文件中出现的顺序排列。如果要进一步操作(如变异检测、IGV查看比对结果等),都需要对BAM文件进行排序,以使比对结果陈列是按“基因组顺序”进行,即根据它们在每个染色体上的位置进行排序。
3.1.1 参数
Usage: samtools sort [options...] [in.bam]
Options:
-l INT Set compression level, from 0 (uncompressed) to 9 (best)
-m INT Set maximum memory per thread; suffix K/M/G recognized [768M]
-n Sort by read name
-t TAG Sort by value of TAG. Uses position as secondary index (or read name if -n is set)
-o FILE Write final output to FILE rather than standard output
-T PREFIX Write temporary files to PREFIX.nnnn.bam
--input-fmt-option OPT[=VAL]
Specify a single input file format option in the form
of OPTION or OPTION=VALUE
-O, --output-fmt FORMAT[,OPT[=VAL]]...
Specify output format (SAM, BAM, CRAM)
--output-fmt-option OPT[=VAL]
Specify a single output file format option in the form
of OPTION or OPTION=VALUE
--reference FILE
Reference sequence FASTA FILE [null]
-@, --threads INT
Number of additional threads to use [0]
3.1.2 参数详解/举例
samtools sort [-l level] [-m maxMem] [-o out.bam] [-O format] [-n] [-T tmpprefix] [-@ threads] [in.sam|in.bam|in.cram]
参数:
-l INT
设置输出文件压缩等级。0-9,0是不压缩,9是压缩等级最高。不设置此参数时,使用默认压缩等级;
-m INT
设置每个线程运行时的内存大小,可以使用K,M和G表示内存大小。
()参数默认下是 500,000,000 即500M(不支持K,M,G等缩写)。对于处理大数据时,如果内存够用,则设置大点的值,以节约时间。
-n
设定排序方式按short reads的ID排序。否则,默认下是按序列在fasta文件中的顺序(即header)和序列从左往右的位点排序。
(1)默认方式,按照染色体的位置进行排序
(2)参数-n则是根据read名进行排序
-o FILE
设置最终排序后的输出文件名;
-O FORMAT
设置最终输出的文件格式,可以是bam,sam或者cram,默认为bam;
-T PREFIX
设置临时文件的前缀;
-@ INT
设置排序和压缩是的线程数量,默认是单线程。
例子:
samtools sort abc.bam abc.sort
# 注意 abc.sort 是输出文件的前缀,实际输出是 abc.sort.bam
samtools sort aln.bam -o aln.sort.bam
3.2 mpileup
输入BAM文件,产生bcf或pileup格式的文件,bcf格式文件可以通过bcftools处理检测snp和indel。Pileup文件可通过varscan检测变异(如snp、indel等)
samtools mpileup test.sort.dup.bam -o test.sort.dup.bcf
# bcftools
bcftools call -Ovm -o out.vcf test.sort.dup.bcf
# samtools+ VarScan
samtools mpileup -f ref.fa test.sort.dup.bam | java -jar VarScan.jar mpileup2snp --output-vcf --strand-filter 0
samtools mpileup -f ref.fa test.sort.dup.bam | java -jar VarScan.jar mpileup2indel --output-vcf --strand-filter 0
samtools mpileup -q20 -d8000 -f ref.fa test.sorted.dup.bam | java -jar VarScan.v2.3.9.jar mpileup2cns --variants --output-vcf > out.vcf
4. 统计(statistics)
4.1 flagstat(BAM文件的简单统计)
4.1.1 参数
Usage: samtools flagstat [options] <in.bam>
--input-fmt-option OPT[=VAL]
Specify a single input file format option in the form
of OPTION or OPTION=VALUE
-@, --threads INT
Number of additional threads to use [0]
4.1.2 举例
samtools flagstat -@ 10 t1.sort.bam > t1.flagstat.txt # @代表线程数
6874858 + 0 in total (QC-passed reads + QC-failed reads)
90281 + 0 duplicates
6683299 + 0 mapped (97.21%)
6816083 + 0 paired in sequencing
3408650 + 0 read1
3407433 + 0 read2
6348470 + 0 properly paired (93.14No value assigned)
6432965 + 0 with itself and mate mapped
191559 + 0 singletons (2.81No value assigned)
57057 + 0 with mate mapped to a different chr
45762 + 0 with mate mapped to a different chr (mapQ>=5)
各列的意义可参考:https://www.jianshu.com/p/ccc59b459d4a
4.1.3 结果解释
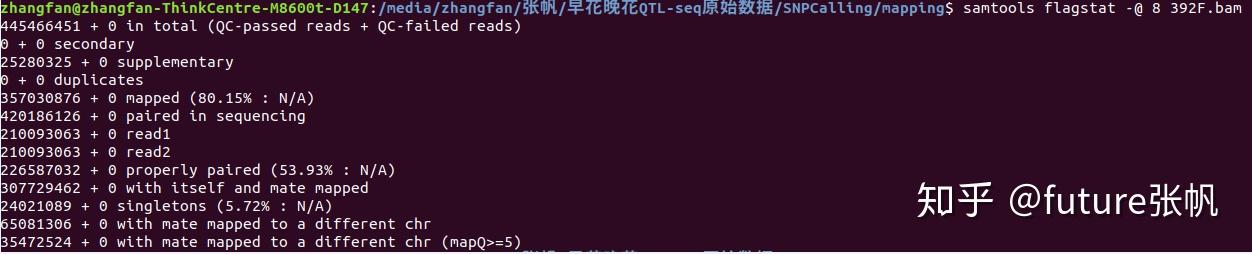
445466451 + 0 in total (QC-passed reads + QC-failed reads)
#质控通过的reads数+质控未通过的reads数,0x200 bit set 后面都是用这个参数区分的
0 + 0 secondary
#次级read数,0x100 bit set (不是很懂怎么算的)
25280325 + 0 supplementary
#额外reads数, 0x800 bit set
0 + 0 duplicates
#重复reads数,0x400 bit set
357030876 + 0 mapped (80.15% : N/A)
#匹配率,这个值比较重要,0x4 bit not set
420186126 + 0 paired in sequencing
#成对的序列数量,0x1 bit set
210093063 + 0 read1
#read1的数量,both 0x1 and 0x40 bits set
210093063 + 0 read2
#read2的数量,both 0x1 and 0x80 bits set
226587032 + 0 properly paired (53.93% : N/A)
#唯一匹配的reads数量,both 0x1 and 0x2 bits set and 0x4 bit not set
#正确匹配的reads数:比对到同一条参考序列,并且两条reads之间的距离符合设置的阈值
307729462 + 0 with itself and mate mapped
#非唯一匹配的reads数量,0x1 bit set and neither 0x4 nor 0x8 bits set
#paired reads中两条都比对到参考序列上的reads数
24021089 + 0 singletons (5.72% : N/A)
#只有一条染色体匹配上参考基因组的数量,both 0x1 and 0x8 bits set and bit 0x4 not set
#单独一条匹配到参考序列上的reads数,和上一个相加,则是总的匹配上的reads数
65081306 + 0 with mate mapped to a different chr
#比对到不同染色体的reads数,0x1 bit set and neither 0x4 nor 0x8 bits set and MRNM not equal to RNAME
#paired reads中两条分别比对到两条不同的参考序列的reads数
35472524 + 0 with mate mapped to a different chr (mapQ>=5)
#比对到不同染色体,但是比对质量值大于5的reads数,0x1 bit set and neither 0x4 nor 0x8 bits set and MRNM not equal to RNAME and MAPQ >= 5
#同上一个,只是其中比对质量>=5的reads的数量
4.2 depth(统计每个位置碱基的测序深度)
samtools depth SRR2584866.aligned.sorted.bam > depth.txt
#result
head depth.txt
CP000819.1 1 4
CP000819.1 2 4
CP000819.1 3 5
CP000819.1 4 5
CP000819.1 5 5
CP000819.1 6 5
CP000819.1 7 5
CP000819.1 8 5
CP000819.1 9 5
CP000819.1 10 5
3列依次为染色体名称,位置,覆盖深度。
4.3 stats
5.Samtools手册
http://www.htslib.org/doc/#manual-pages
http://www.htslib.org/doc/samtools.html