07重测序流程1-常规有参
〇. 一个项目出多份报告,看此节,否则往下看
## 在项目编号目录下,出几份报告就创建几个对应文件夹
1.显示$i文件夹
for i in `ls`;do cd $i && echo `ls` && cd -;done
2.查找染色体,默认选择前15个
for i in `ls`;do cd $i/data && pwd && ir.py -i *.fna -l | sort -k 1 -nr > len.txt && cat len.txt | head -n 15 | awk '{print $2}' > chrlist.txt && cat chrlist.txt && cd -;done
3.提取cds
for i in `ls`;do cd $i/data && gffread -x cds.fa -g *.fna *.gff && perl /share/nas6/zhouxy/functional_modules/cnvfmt_trans2longest/fasta2longest_with_gff3.pl -i cds.fa -g *.gff -o filter.cds.fa && cp /share/nas6/pub/pipline/gene-function-annotation/v1.3/profile/fungi_QAll.cfg ./ && cd - & done
4.填写filter.cds.fa绝对路径
## 真菌只改绝对路径
for i in `ls`;do cd $i/data && ll fungi_QAll.cfg && cd - & done
path=`pwd` && for i in `ls`; do echo "sed -i '1s|/share/nas6/zhanghk/project/ref/GP-20210708-3193_jiangnandaxue_12samples_jiaomu_trans/CBS7435/cbs7435_CDS.fa|"$path"/"$i"/data/filter.cds.fa|' "$path"/"$i"/data/fungi_QAll.cfg" >> modified1.sh;done && sh modified1.sh # 批量填写路径
5.功能注释
for i in `ls`;do cd $i/data && nohup perl /share/nas6/pub/pipline/gene-function-annotation/v1.3/Gene_Func_Anno_Pipline.pl --nr --swissprot --cog --kog --kegg --pfam --GO --cfg *_QAll.cfg --od Basic_function & done
6.生成配置文件
for i in `ls`;do cd $i && perl /share/nas6/xul/program/reseq/create_profile_for_reseq.pl -i 下发路径/2.clean_data/*/ -s data/*.fna -g data/chrlist.txt -gff data/*.gff -p $i -c data/Basic_function/Result/All_Database_annotation.xls && cd - & done
7.修改配置文件,运行主流程
for i in `ls`;do cd $i && nohup perl /share/nas1/yuj/pipline/reseq/v1.2/re_seq.pl -i ref_reseq_config.yaml -o analysis_dir & done
一.数据准备
Info
主要是准备4.染色体列表,通常其他信息都有。
查找基因组看“8️⃣.查找参考基因组”
1. 测序所在路径,一般有多个文件夹,每个样品一个文件夹
2. 基因组fasta文件,有时需要下载,有时客户提供,还有常见的或者我们做过的可以到 /share/nas6/pub/genome中找找
gzip -d file.gz
gunzip file.gz
3. 基因组注释文件,gff3格式
## gbk转gff
perl /share/nas6/xul/program/chloroplast/mvista/genbank.to.gff.pl -i *.gbk -f *.fasta -o tmp.gff
perl -lane '/Name=(.*;)/;print $_."Parent=$1ID=$1"' tmp.gff > genome.gff #修改名字
4. 染色体列表,txt文件,一列
存放序列名称,因为有的fasta文件中有很多序列,该文件用于定义某些图片中需要展示的序列。
一般是保留染色体级别的序列,可以保留所有染色体
如果该基因组都是scaffold,那么就保留几个比较长的序列名称,十个左右就行
ir.py -i *.fna -l | sort -k 1 -nr > len.txt
cat len.txt | head -n 15 | awk '{print $2}' > chrlist.txt && cat chrlist.txt # 根据情况取前多少行
5. 功能注释结果的整合文件
二.功能注释 --CDS版
可放后台 如果已经做过了,可以跳过该步骤,nas6中的基因组一般都做过这个分析 /share/nas6/pub/genome
2.0 合并步骤
通常看这个就行,后面几步是分步来做
data]$
1.挑选cds
gffread -x cds.fa -g *.fna *.gff && perl /share/nas1/yuj/functional_modules/cnvfmt_trans2longest/fasta2longest_with_gff3.pl -i cds.fa -g *.gff -o filter.cds.fa
2.填写配置
# 原路径 /share/nas6/pub/pipline/gene-function-annotation/v1.3/profile
新路径 /share/nas1/yuj/pipline/gene-function-annotation/v1.3/profile
## 真菌
cp /share/nas1/yuj/pipline/gene-function-annotation/v1.3/profile/fungi_QAll.cfg ./
path=`pwd` && sed -i "1s|/share/nas6/zhanghk/project/ref/GP-20210708-3193_jiangnandaxue_12samples_jiaomu_trans/CBS7435/cbs7435_CDS.fa|$path/filter.cds.fa|" $path/fungi_QAll.cfg
## 细菌
cp /share/nas1/yuj/pipline/gene-function-annotation/v1.3/profile/BCT_QAll.cfg ./
path=`pwd` && sed -i "1s|/share/nas6/yangl/project/2022/drna_seq/GP-20220113-3957_neinongda_2samples_xujun_trans/genomic/sequence.cds.fa|$path/filter.cds.fa|" $path/BCT_QAll.cfg
## 双子叶植物
cp /share/nas1/yuj/pipline/gene-function-annotation/v1.3/profile/plant_QAll.cfg ./
path=`pwd` && sed -i "1s|/share/nas6/zhail/project/denoref/202106/GP-20210423-2913_shenyangnongye_fengxin_6samples_juhua_trans/analysis_dir/trinity_out_dir/Unigenes.fasta|$path/filter.cds.fa|" $path/plant_QAll.cfg
All_QAll.cfg
BCT_QAll.cfg #细菌只改绝对路径
danziye_plant_QAll.cfg
fungi_QAll.cfg #真菌只改绝对路径
Gene_Func_Anno_Pipline.cfg
INV_QAll.cfg # 无脊椎动物(Invertebrate)
MAM_QAll.cfg # 哺乳动物(Mammal)
plant_QAll.cfg #植物改“绝对路径”“NR”“KEGG”,双子叶植物项目只改路径
ROD_QAll.cfg # 啮齿动物(Rodent)
VRL_QAll.cfg # 病毒(Virus)
VRT_QAll.cfg # 脊椎动物(Vertebrate)
3.功能注释
nohup perl /share/nas6/pub/pipline/gene-function-annotation/v1.3/Gene_Func_Anno_Pipline.pl --nr --swissprot --cog --kog --kegg --pfam --GO --cfg *_QAll.cfg --od Basic_function &
2.1 获取转录本序列文件
1. 获取转录本序列文件,fasta格式的核酸序列
$ gffread -x cds.fa -g *.fna *.gff
# 遇到错误就删删删 如下
$ grep -n "rna-A564_p001" GCF_004118075.2_ASM411807v2_genomic.gff | wc -l
$ for i in {1..6};do echo $i;sed -i '649514d' GCF_004118075.2_ASM411807v2_genomic.gff;done
# cds要用id来命名,重测序这块分析可能不用管
2.2 获取最长转录本
2.获取最长转录本
有的基因可能存在多个转录本,所以需要过滤下,根据上述提取出来的基因名称判断哪些是同一个基因的,一般id后面有.1 .2这种,就属于一个基因的不同转录本。
具体还要看提取出来的格式。
# perl /share/nas6/zhouxy/functional_modules/cnvfmt_trans2longest/fasta2longest.pl -i cds.fa -o filter.cds.fa # 旧的程序不用 不使用gff,不会改变序列id
$ perl /share/nas6/zhouxy/functional_modules/cnvfmt_trans2longest/fasta2longest_with_gff3.pl -i cds.fa -g *.gff -o filter.cds.fa # 用这个 会把rna的id替换成所属gene的id
$ perl /share/nas1/yuj/functional_modules/cnvfmt_trans2longest/fasta2longest_with_gff3.pl -i cds.fa -g *.gff -o filter.cds.fa # 输出id映射,后面富集分析记得改id
# perl /share/nas6/zhouxy/functional_modules/cnvfmt_trans2longest/fasta_trans_to_geneid_format_convert_ncbi.pl 格式转换 没用过
2.3 填写功能注释的配置文件
3. 填写功能注释的配置文件
## 植物 下面的nr库需要根据情况改成单子叶的或者双子叶的,默认双子叶
$ cp /share/nas1/yuj/pipline/gene-function-annotation/v1.3/profile/plant_QAll.cfg ./
## 病毒
$ cp /share/nas1/yuj/pipline/gene-function-annotation/v1.3/profile/VRL_QAll.cfg ./
##真菌只改绝对路径
$ cp /share/nas1/yuj/pipline/gene-function-annotation/v1.3/profile/fungi_QAll.cfg ./
path=`pwd` && sed -i "1s|/share/nas6/zhanghk/project/ref/GP-20210708-3193_jiangnandaxue_12samples_jiaomu_trans/CBS7435/cbs7435_CDS.fa|$path/filter.cds.fa|" $path/fungi_QAll.cfg
修改第一行,改成filter.cds.fa的绝对路径!
2.4 运行功能注释
4. 运行功能注释
nohup perl /share/nas6/pub/pipline/gene-function-annotation/v1.3/Gene_Func_Anno_Pipline.pl --nr --swissprot --cog --kog --kegg --pfam --GO --cfg *_QAll.cfg --od Basic_function &
程序运行完后需要用到的文件就是:Basic_function/Result/All_Database_annotation.xls 这个文件,用于填写下面的配置文件
2.5 整合程序未运行的cog注释
## cog注释
perl /share/nas6/pub/pipline/gene-function-annotation/v1.3/bin/Gene_Cog_Anno.pl -id /share/nas1/yuj/project/re-sequencing/GP-20230327-5799_20230519/202309-groupD/data/Basic_function/mid -Database /share/nas6/pub/database/cog/v20180610/myva -od /share/nas1/yuj/project/re-sequencing/GP-20230327-5799_20230519/202309-groupD/data/Basic_function -Blast_cpu 80 -Blast_e 1e-5
## 整合进最终的xls文件
perl /share/nas6/pub/pipline/gene-function-annotation/v1.3/bin/anno_integrate.pl -gene /share/nas1/yuj/project/re-sequencing/GP-20230327-5799_20230519/202309-groupD/data/filter.cds.fa -id /share/nas1/yuj/project/re-sequencing/GP-20230327-5799_20230519/202309-groupD/data/Basic_function/Result -od /share/nas1/yuj/project/re-sequencing/GP-20230327-5799_20230519/202309-groupD/data/Basic_function/Result -key filter.cds.fa
二.功能注释 -- 蛋白版
可放后台 如果已经做过了,可以跳过该步骤,nas6中的基因组一般都做过这个分析 /share/nas6/pub/genome
data]$
1.挑选cds
gffread -x cds.fa -g *.fna *.gff && perl /share/nas1/yuj/functional_modules/cnvfmt_trans2longest/fasta2longest_with_gff3.pl -i cds.fa -g *.gff -o filter.cds.fa && ir.py -s7 -n 1 -i filter.cds.fa -o filter.cds.pep
2.填写配置
第一行mrna用不到,不用管
## 细菌
cp /share/nas1/yuj/pipline/gene-function-annotation/v1.3/profile/BCT_QAll.cfg ./
## 真菌
cp /share/nas1/yuj/pipline/gene-function-annotation/v1.3/profile/fungi_QAll.cfg ./
## 双子叶
cp /share/nas1/yuj/pipline/gene-function-annotation/v1.3/profile/plant_QAll.cfg ./
All_QAll.cfg
BCT_QAll.cfg
danziye_plant_QAll.cfg
fungi_QAll.cfg
Gene_Func_Anno_Pipline.cfg
INV_QAll.cfg # 无脊椎动物(Invertebrate)
MAM_QAll.cfg
plant_QAll.cfg # 单子叶改NR/KEGG数据库,双子叶不用改 /share/nas6/pub/database/nr/v20200108/PLANT/DZY.pep.fa
ROD_QAll.cfg
VRL_QAll.cfg
VRT_QAll.cfg
3.功能注释
# nohup perl /share/nas6/pub/pipline/prot-function-annotation/current/Gene_Func_Anno_Pipline_v2.pl --all --cfg *_QAll.cfg --od Basic_function -fa filter.cds.pep &
nohup perl /share/nas2/pub/pipline/prot-function-annotation/v1.3/Gene_Func_Anno_Pipline_v2.pl --all --cfg *_QAll.cfg --od Basic_function -fa filter.cds.pep &
三.有参流程
3.1 配置文件(v1.3流程需要分组文件)
配置模板:
/share/nas6/zhouxy/project/reseq/GP-20210820-3319_jiangsunongkeyuannongchanpinjiagongsuo_1samples_xijun/re_config.yaml
程序参数:
perl /share/nas6/xul/program/reseq/create_profile_for_reseq.pl
-i <in dir> contain all.gz file or fq or fastq
# 输入包含测序文件所在的路径(文件夹,不是具体文件路径),如 path/clean/*/
-s ref genome
# 输入参考基因组fasta文件
-g ref chr list
# 输入染色体列表
-c ref anno Gene_Anno/Result/All_Database_annotation.xls default
# 输入功能注释的结果Basic_function/Result/All_Database_annotation.xls,如果功能注释没做完,这个可以先不填。
-gff ref gff
# 输入gff文件
-Is insert_size,default 100-500
# 插入片段大小,一般不用改
-p Prefix default XM
# 物种名简拼,不改也行
应用(看这)
cd ../
datapath=下发路径
for i in `ls $datapath/2.clean_data | grep -v txt`;do echo -e "$i\tALL";done > sample.group.txt
perl /share/nas6/xul/program/reseq/create_profile_for_reseq.pl -s data/*.fna -g data/chr*.* -gff data/*.gff* -c data/Basic_function/Result/All_Database_annotation.xls -p BY -i $datapath/2.clean_data/*/
# -i可以只输入制定样品 -i 2.clean_data/D1110/
echo -e "\n\tGroups : $(realpath sample.group.txt)" >> ref_reseq_config.yaml
3.2 运行主程序
/share/nas6/zhouxy/pipline/reseq/v1.3/re_seq.pl
/share/nas2/zhouxy/pipline2/reseq/v1.3/re_seq.pl
/share/nas1/yuj/pipline/reseq/v1.3/re_seq.pl
nohup perl /share/nas1/yuj/pipline/reseq/v1.3/re_seq.pl -i ref_reseq_config.yaml -o analysis_dir &
3.3 整理结果
# perl /share/nas1/yuj/pipline/reseq/v1.2/script/resultdir/resultDir.pl -i analysis_dir -o complete_dir &
# 去除了06里的cmds
# sh analysis_dir/.cmds_dir/genomeStat.cmds 有时候统计信息没生成
perl /share/nas1/yuj/pipline/reseq/v1.3/script/resultdir/resultDir.pl -i analysis_dir -o complete_dir &
3.4 生成报告
1.修改配置文件
# /share/nas6/zhouxy/pipline/reseq/current/re-report_html.cfg
cp /share/nas2/yuj/project/2024/re-sequencing/GP-20231007-6973_20240227/3/report.cfg ./report.cfg && realpath ./report.cfg
2.生成报告
# perl /share/nas6/zhouxy/pipline/reseq/current/re-report_html_for_lumpy_sv.pl -id complete_dir/ -cfg ./report.cfg # 有点小问题,先不用
# 售后QQ:2893941692
# perl /share/nas1/yuj/pipline/reseq/v1.2/re-report_html_for_lumpy_sv.pl -id complete_dir/ -cfg ./report.cfg
perl /share/nas1/yuj/pipline/reseq/v1.3/re-report_html_for_lumpy_sv.pl -id complete_dir/ -cfg ./report.cfg
-- 20230928 当基因功能注释文件夹不存在时去除报告相关内容
-- 20240426 当染色体结构变异文件夹不存在时去除报告相关内容
四.没有GFF时
舍弃一系列的indel部分
去掉某一步的rscript画图(可能不需要)
1.以下indel部分无法生成,会被过滤掉,跳过即可
touch analysis_dir/.flag_dir/gatk_VQSR.ok
touch analysis_dir/.flag_dir/gatk_VariantFiltration.ok
继续跑
2.跳过6.1 snpeff_index.ok这一步
可以不用管
3.在下一步注释,直接复制就行
cp analysis_dir/variation_dir/variants_anno_dir/samples.gatk.con.snp.vqsr.filter.vcf analysis_dir/variation_dir/variants_anno_dir/samples.gatk.con.snp.vqsr.filter.anno.vcf
4.运行Variant_Annotation.cmds的第2/3条命令
5.gatk_MergeVcfs.cmds去掉indel合并后运行
sh analysis_dir/.cmds_dir/gatk_MergeVcfs.cmds
touch analysis_dir/.flag_dir/gatk_MergeVcfs.ok
继续跑
6.diff_Variation.cmds去掉indel部分后运行
sh analysis_dir/.cmds_dir/diff_Variation.cmds
touch analysis_dir/.flag_dir/diff_Variation.ok
继续跑
7.analysis_dir/.cmds_dir/vcf_filter.cmds也会报错
不用管,能生成ok文件
8.popSNP_Stat.cmds去掉indel部分后运行
sh analysis_dir/.cmds_dir/popSNP_Stat.cmds
touch analysis_dir/.flag_dir/popSNP_Stat.ok
继续跑
五.出现的问题(v1.3)
5.1 SV无结果
ll analysis_dir/SV/groups_detect # 里面是空文件
重新运行即可
sh analysis_dir/.cmds_dir/all.SVdetect.cmds &
5.2 mapping_dir图片(真菌项目)
5.2.1 bam2depth
1.bam2depth.cmds
depth文件大小差别大,是因为没有输出0位置
samtools depth -m 100000 -a # 最大值100000,输出0位置
touch analysis_dir/.flag_dir/bam2depth.ok
5.2.2 深度窗口绘图
2.depth2windows.cmds
真菌染色体短,窗口设置成1k
-d 1000
touch analysis_dir/.flag_dir/depth2windows.ok
3.绘制轨道图(注意深度)
sh analysis_dir/.cmds_dir/depth_Track_point.cmds
touch analysis_dir/.flag_dir/depth_Track_point.ok
5.2.3 mapping_stat/bcparse
3.bam2bc_stat.cmds samples_mapping_stat.cmds
见v1.2的5.2.1
先生成bc,再重新绘图
for i in {BY4741,FG2-QC,XNB};do /share/nas1/yuj/pipline/reseq/v1.3/script/01bam/coverage_depth.R --i $i.coverage.csv --o $i.coverage;done
5.3 富集分析没有图(同v1.2版本)
5.4 snpeff运行缓慢 Variant_Annotation.cmds
换一个版本来跑这一步
/share/nas6/zhouxy/software/java/current/bin/java -jar /share/nas6/yangl/biosoft/snpEff/snpEff_v3_6/snpEff.jar MH -o gatk -ud 2000 -csvStats -s /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.snp.stat.csv -s /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.snp.stat.html -c /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/snpeff_index/MH.config -v /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.gatk.con.snp.vqsr.filter.vcf > /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.gatk.con.snp.vqsr.filter.anno.vcf
/share/nas6/zhouxy/biosoft/perl/current/bin/perl /share/nas1/yuj/pipline/reseq/v1.3/script/02variant/extract_oneEff_anno.pl -i /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.gatk.con.snp.vqsr.filter.anno.vcf -o /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.pop.snp.anno.result.vcf
/share/nas6/zhouxy/biosoft/perl/current/bin/perl /share/nas1/yuj/pipline/reseq/v1.3/script/02variant/vcf_to_snplist_v1.4.pl -i /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.pop.snp.anno.result.vcf -o /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.pop.snp.anno.result.list -ref 1
## indel
/share/nas6/zhouxy/software/java/current/bin/java -jar /share/nas6/yangl/biosoft/snpEff/snpEff_v3_6/snpEff.jar MH -o gatk -ud 2000 -csvStats -s -s/share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.indel.stat.csv -s /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.indel.stat.html -c /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/snpeff_index/MH.config -v /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.gatk.con.indel.vqsr.filter.vcf > /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.gatk.con.indel.vqsr.filter.anno.vcf
/share/nas6/zhouxy/biosoft/perl/current/bin/perl /share/nas1/yuj/pipline/reseq/v1.3/script/02variant/extract_oneEff_anno.pl -i /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.gatk.con.indel.vqsr.filter.anno.vcf -o /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.pop.indel.anno.result.vcf
/share/nas6/zhouxy/biosoft/perl/current/bin/perl /share/nas1/yuj/pipline/reseq/v1.3/script/02variant/vcf_to_indellist_v1.5.pl -i /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.pop.indel.anno.result.vcf -o /share/nas2/yuj/project/2025/re-sequencing/GP-20241021-9521-2_20250104/analysis_dir/variation_dir/variants_anno_dir/samples.pop.indel.anno.result.list -ref 1
五.检查结果(v1.2)
5.1 SV无结果
ll analysis_dir/SV/groups_detect # 里面是空文件
重新运行即可
sh analysis_dir/.cmds_dir/all.SVdetect.cmds &
5.2 bcparse 测序深度/插入片段
5.2.0 重新运行
通常重新运行samples_align_stats.cmds即可
但有时候这个cmd文件未生成,
需要运行下面命令
perl /share/nas1/yuj/pipline/reseq/v1.2/script/01bam/samtools_stats_parser2.pl -i analysis_dir/mapping_dir/mapping_stat/bcfiles -o analysis_dir/mapping_dir/mapping_stat/bcparse -r analysis_dir/genome/*.fna
display analysis_dir/mapping_dir/mapping_stat/bcparse/*.coverage.png
display analysis_dir/mapping_dir/mapping_stat/bcparse/*.insertsize.png
5.2.1 Q1:测序深度太高
分为统计和绘图两部分,优先尝试仅更改绘图参数
1.
cd analysis_dir/mapping_dir/mapping_stat/bcparse
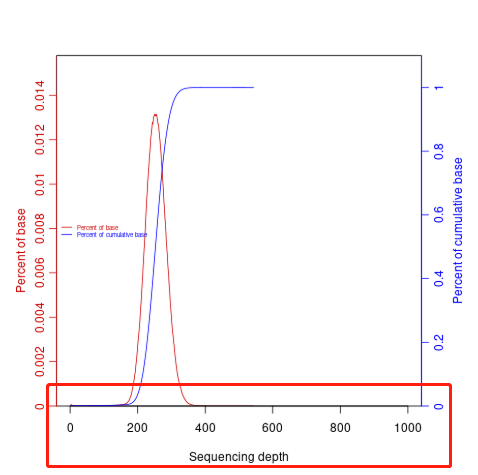
2.根据*.coverage.csv深度来调整else分支的最大值,其控制的是下图中的横坐标最大值
/share/nas1/yuj/pipline/reseq/v1.2/script/01bam/coverage_depth.R 42行
if (xmean < 10){xmax=20;}else{xmax=200} # 默认为200,深度高就改成1000
3.然后先尝试一次画图
/share/nas1/yuj/pipline/reseq/v1.2/script/01bam/coverage_depth.R --i *.coverage.csv --o $sample.coverage
4.不行的话再看下面从头调整文件然后画图
下面是尝试不断更改统计参数
参数说明:
-c, --coverage <int>,<int>,<int>:覆盖度分布的最小值、最大值和步长,默认为1,1000,1
1)遇到细菌项目,可以 -c 1,10000000,1
然后根据实际情况再去更改上图的xmax
2)以下都是某个真菌项目,更改的是步长参数,xmax参数未改
## 单个样品for循环 step从100画到2900
analysis_dir/mapping_dir/01.bwa2sortbam]$
for i in {1..29};do samtools stats -c 1,50000,$i"00" EFV1.sort.bam > ../mapping_stat/bcfiles/$i"00EFV1.bc" && grep ^COV ../mapping_stat/bcfiles/$i"00EFV1.bc" | cut -f 3- > ../mapping_stat/bcparse/$i"00EFV1.coverage.csv" && cd ../mapping_stat/bcparse/ && /share/nas1/yuj/pipline/reseq/v1.2/script/01bam/coverage_depth.R --i $i"00EFV1.coverage.csv" --o $i"00EFV1.coverage" && cd - & done
## 固定参数 for循环 挨个样品画
for i in {Del-E,Del-L,FAdV-4,H9-HA-E,H9-HA-L,rF2}; do samtools stats -c 1,50000,1500 $i.sort.bam | grep ^COV | cut -f 3- > ../mapping_stat/bcparse/$i.coverage.csv && cd ../mapping_stat/bcparse/ && /share/nas1/yuj/pipline/reseq/v1.2/script/01bam/coverage_depth.R --i $i.coverage.csv --o $i.coverage && cd - ;done
## 单样品画图 手动修改参数
samtools stats -c 1,500000,1 EFV1.sort.bam > ../mapping_stat/bcfiles/EFV1.bc && grep ^COV ../mapping_stat/bcfiles/EFV1.bc | cut -f 3- > ../mapping_stat/bcparse/EFV1.coverage.csv && cd ../mapping_stat/bcparse/ && /share/nas1/yuj/pipline/reseq/v1.2/script/01bam/coverage_depth.R --i EFV1.coverage.csv --o EFV1.coverage && cd -
5.2.2 Q2:插入片段重影
1.R
$ Rscript /share/nas1/yuj/pipline/reseq/v1.2/script/01bam/insert_size.R --i I4.insertsize.csv --o I4.insertsize # 原流程存在重影问题
2.python
python3 /share/nas1/yuj/pipline/reseq/v1.2/script/01bam/insert_size.R.py -i *.insertsize.csv -o $sample.insertsize # 修复了重影问题
5.3 富集分析没有图
display analysis_dir/variant_gene/snp.variant_gene_Annotation/GO/snp.variant_gene.GO.png
其实就是让analysis_dir/variant_gene/xxx.variant_gene.ID文件第一列和data/Basic_function/Result/All_Database_annotation.xls的第一列完全一致
5.3.1 id文件缺“gene-”前缀
发现id文件缺“gene-”前缀可以直接看这一步,一股脑全部运行就OK
## 给过滤的vcf添加功能注释
perl /share/nas1/yuj/pipline/reseq/v1.2/script/05vcf_stat/region_info_filterd.pl -i data/Basic_function/Result/All_Database_annotation.xls -s analysis_dir/vcf_filter/samples.pop.snp.filter.list -o analysis_dir/vcf_filter/samples.pop.snp.filter.anno.xls &
perl /share/nas1/yuj/pipline/reseq/v1.2/script/05vcf_stat/region_info_filterd.pl -i data/Basic_function/Result/All_Database_annotation.xls -s analysis_dir/vcf_filter/samples.pop.indel.filter.list -o analysis_dir/vcf_filter/samples.pop.indel.filter.anno.xls &
## 修改两个id文件
tmp=`sed -n "2p" analysis_dir/variant_gene/snp.variant_gene.ID | awk -F "_" '{print $1}'` && sed -i "s/$tmp/gene-$tmp/g" analysis_dir/variant_gene/snp.variant_gene.ID && sed -i "s/$tmp/gene-$tmp/g" analysis_dir/variant_gene/indel.variant_gene.ID
## 运行富集分析
sh /share/nas1/yuj/pipline/reseq/v1.2/script/07DEG_Anno/DEG_Anno.sh analysis_dir/variant_gene/snp.variant_gene.ID snp.variant_gene analysis_dir/variant_gene data/Basic_function/Result/All_Database_annotation.xls && perl /share/nas1/yuj/pipline/reseq/v1.2/script/07DEG_Anno/anno.stat.pl -i analysis_dir/variant_gene -o analysis_dir/variant_gene/anno.stat.xls &
sh /share/nas1/yuj/pipline/reseq/v1.2/script/07DEG_Anno/DEG_Anno.sh analysis_dir/variant_gene/indel.variant_gene.ID indel.variant_gene analysis_dir/variant_gene data/Basic_function/Result/All_Database_annotation.xls && perl /share/nas1/yuj/pipline/reseq/v1.2/script/07DEG_Anno/anno.stat.pl -i analysis_dir/variant_gene -o analysis_dir/variant_gene/anno.stat.xls &
## 上面所有命令都写入了该文件,一般不用这个sh脚本
sh /share/nas1/yuj/pipline/reseq/v1.2/script/variant_gene.sh
5.3.2 分步运行
1.vcf_filter.cmds里面snp/indel的最后一部分重新运行
# /share/nas6/zhouxy/biosoft/perl/current/bin/perl /share/nas1/yuj/pipline/reseq/v1.2/script/05vcf_stat/region_info_filterd.pl
# 放后台就行
perl /share/nas1/yuj/pipline/reseq/v1.2/script/05vcf_stat/region_info_filterd.pl -i data/Basic_function/Result/All_Database_annotation.xls -s analysis_dir/vcf_filter/samples.pop.snp.filter.list -o analysis_dir/vcf_filter/samples.pop.snp.filter.anno.xls &
perl /share/nas1/yuj/pipline/reseq/v1.2/script/05vcf_stat/region_info_filterd.pl -i data/Basic_function/Result/All_Database_annotation.xls -s analysis_dir/vcf_filter/samples.pop.indel.filter.list -o analysis_dir/vcf_filter/samples.pop.indel.filter.anno.xls &
2.生成mapping.id
cd data && perl /share/nas1/yuj/functional_modules/cnvfmt_trans2longest/fasta2longest_with_gff3.pl -i cds.fa -g *.gff -o tmp && cp mapping.id ../analysis_dir/variant_gene/ && cd ../analysis_dir/variant_gene/ && cd ../../ # 输出id映射
3.variant_gene.cmds里面运行snp/indel第一部分生成ID文件
perl /share/nas1/yuj/pipline/reseq/v1.2/script/06varGene/get_gene_info.pl -i analysis_dir/vcf_filter/samples.pop.snp.filter.list -o analysis_dir/variant_gene/snp.variant_gene.ID &
perl /share/nas1/yuj/pipline/reseq/v1.2/script/06varGene/get_gene_info.pl -i analysis_dir/vcf_filter/samples.pop.indel.filter.list -o analysis_dir/variant_gene/indel.variant_gene.ID &
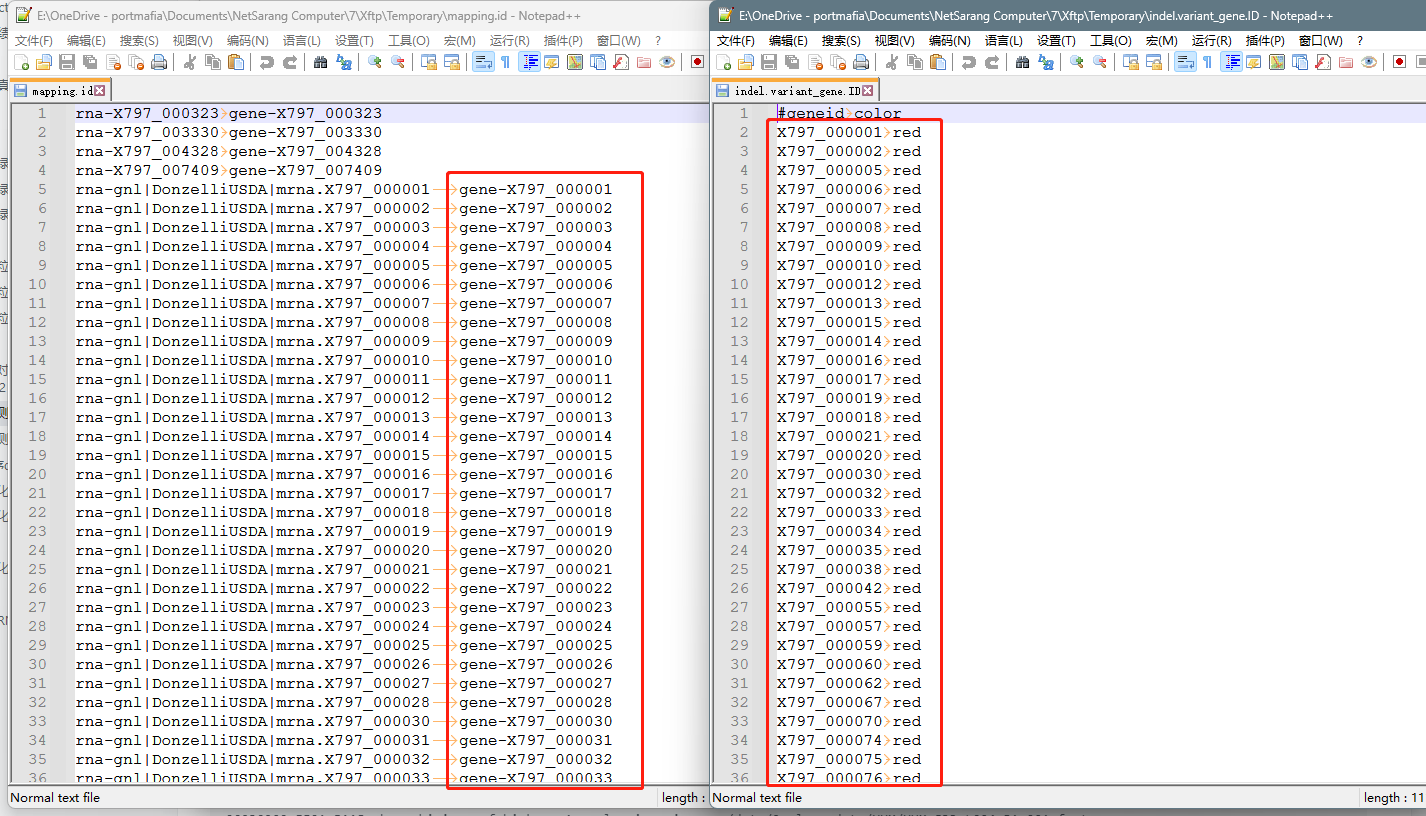
4.修改ID文件里的前缀,再运行替换命令
4.1 如果差别仅仅只是gene-,如上图所示,右侧框里少了gene-,不用awk来替换,直接文本编辑就ok,4.2的替换命令可以跳过

4.2 但如果是上图所示,就需要手动先把右侧文件编辑成左边框的格式,然后运行下面的命令
# 注意分隔用制表符
analysis_dir/variant_gene]
$ awk 'BEGIN{OFS="\t"} NR==FNR{mapping[$1]=$2; next} $1 in mapping{$1=mapping[$1]} 1' mapping.id indel.variant_gene.ID > indel.variant_gene.ID2
$ awk 'BEGIN{OFS="\t"} NR==FNR{mapping[$1]=$2; next} $1 in mapping{$1=mapping[$1]} 1' mapping.id snp.variant_gene.ID > snp.variant_gene.ID2
$ rm *.ID mapping.id && rename ID2 ID *
5.variant_gene.cmds里面snp/indel从sh开始运行
sh /share/nas1/yuj/pipline/reseq/v1.2/script/07DEG_Anno/DEG_Anno.sh analysis_dir/variant_gene/snp.variant_gene.ID snp.variant_gene analysis_dir/variant_gene data/Basic_function/Result/All_Database_annotation.xls && perl /share/nas1/yuj/pipline/reseq/v1.2/script/07DEG_Anno/anno.stat.pl -i analysis_dir/variant_gene -o analysis_dir/variant_gene/anno.stat.xls &
sh /share/nas1/yuj/pipline/reseq/v1.2/script/07DEG_Anno/DEG_Anno.sh analysis_dir/variant_gene/indel.variant_gene.ID indel.variant_gene analysis_dir/variant_gene data/Basic_function/Result/All_Database_annotation.xls && perl /share/nas1/yuj/pipline/reseq/v1.2/script/07DEG_Anno/anno.stat.pl -i analysis_dir/variant_gene -o analysis_dir/variant_gene/anno.stat.xls &
5.4 密度图
针对几万bp长度的染色体
/share/nas1/yuj/pipline/reseq/v1.2/script/03vcf/VariationDraw_kb.pl -b 1k
5.5 mapping_depth图片未生成 通常不管
重测序其实可以不管
原因
会提示id不在输入文件进而无法绘图
另外,发现图形不是平滑曲线,也是参数的问题
换参数重新生成fordraw可以解决
重新运行
## 程序
1.滑窗统计
/share/nas6/zhouxy/biosoft/bin/depth_stat_windows
-i file input file
-o file output file
-w num windows size
-h usage
-w参数默认使用1000000,生成的fordraw如下,第二列都是1
JAKREL010000100.1 1 10.531282
JAKREL010000101.1 1 9.961758
JAKREL010000102.1 1 10.662214
JAKREL010000103.1 1 10.242439
JAKREL010000104.1 1 9.998680
JAKREL010000105.1 1 10.267645
JAKREL010000106.1 1 9.822779
2.r绘图
/share/nas1/yuj/pipline/reseq/v1.2/script/01bam/genomeCoveragehorizontalArea.R
上个程序 -w 1000000 对应绘图程序的 -unit Mb
以真菌如何修改为例(真菌染色体长度较短,导致参数不匹配)
1.修改bam2depth.cmds第二部分的 -w参数值
根据len.txt中最长长度135547,-w设置为1000,也就是以Kb为单位
修改成如下/share/nas6/zhouxy/biosoft/samtools/current/bin/samtools depth /share/nas1/yuj/project/re-sequencing/GP-20230327-5799_20230519/m8/analysis_dir/mapping_dir/01.bwa2sortbam/M8.sort.bam > /share/nas1/yuj/project/re-sequencing/GP-20230327-5799_20230519/m8/analysis_dir/mapping_dir/mapping_depth/M8.depth && /share/nas6/zhouxy/biosoft/bin/depth_stat_windows -i /share/nas1/yuj/project/re-sequencing/GP-20230327-5799_20230519/m8/analysis_dir/mapping_dir/mapping_depth/M8.depth -o /share/nas1/yuj/project/re-sequencing/GP-20230327-5799_20230519/m8/analysis_dir/mapping_dir/mapping_depth/M8.depth.fordraw -w 1000
head data/len.txt && sed -i 's/-w 1000000/-w 1000/g' analysis_dir/.cmds_dir/bam2depth.cmds && sh analysis_dir/.cmds_dir/bam2depth.cmds &
2.genomeCoveragehorizontalArea.cmds修改 --unit为Kb
修改成如下/share/nas6/zhouxy/biosoft/R/current/bin/Rscript /share/nas1/yuj/pipline/reseq/v1.2/script/01bam/genomeCoveragehorizontalArea.R --infile /share/nas1/yuj/project/re-sequencing/GP-20230327-5799_20230519/m8/analysis_dir/mapping_dir/mapping_depth/M8.depth.fordraw --idfile /share/nas1/yuj/project/re-sequencing/GP-20230327-5799_20230519/m8/data/chrlist.txt --outfile /share/nas1/yuj/project/re-sequencing/GP-20230327-5799_20230519/m8/analysis_dir/mapping_dir/mapping_depth/M8.genome.coverage --group.col 1 --x.col 2 --y.col 3 --x.lab Sequence-Position --y.lab AverageDepth-log2 --skip 0 --unit Kb --log2
sed -i 's/--unit Mb/--unit Kb/g' analysis_dir/.cmds_dir/genomeCoveragehorizontalArea.cmds && sh analysis_dir/.cmds_dir/genomeCoveragehorizontalArea.cmds &
5.6 samtools index: failed to create index for "xxx" Numerical result out of range
$ /share/nas1/yuj/software/bwa-mem2-2.2.1_x64-linux/bwa-mem2 mem -M -t 24 -R '@RG\tID:HHM\tLB:HHM\tPL:ILLUMINA\tSM:HHM' /share/nas1/yuj/project/re-sequencing/GP-20230209-5591_20230317/analysis_dir/genome/iwgsc_refseqv2.1_assembly.fa /share/nas1/seqloader/reseq/GP-20230209-5591_JAAS_zhongzhixiyuan_fubisheng_1sample_xiaomai_reseq/data/2.clean_data/HHM/HHM_S33_L004_R1_001.fastq.gz /share/nas1/seqloader/reseq/GP-20230209-5591_JAAS_zhongzhixiyuan_fubisheng_1sample_xiaomai_reseq/data/2.clean_data/HHM/HHM_S33_L004_R2_001.fastq.gz | /share/nas6/zhouxy/biosoft/samtools/current/bin/samtools sort -O bam -@ 4 -m 1G -T /share/nas1/yuj/project/re-sequencing/GP-20230209-5591_20230317/analysis_dir/mapping_dir/01.bwa2sortbam/HHM -o /share/nas1/yuj/project/re-sequencing/GP-20230209-5591_20230317/analysis_dir/mapping_dir/01.bwa2sortbam/HHM.sort.bam - && /share/nas6/zhouxy/biosoft/samtools/current/bin/samtools index /share/nas1/yuj/project/re-sequencing/GP-20230209-5591_20230317/analysis_dir/mapping_dir/01.bwa2sortbam/HHM.sort.bam &
报错信息如下:
[E::hts_idx_push] Region 536870840..536870990 cannot be stored in a bai index. Try using a csi index with min_shift = 14, n_lvls >= 6
samtools index: failed to create index for "/share/nas1/yuj/project/re-sequencing/GP-20230209-5591_20230317/analysis_dir/mapping_dir/01.bwa2sortbam/HHM.sort.bam": Numerical result out of range
解决方法:换用其他版本
$ wget https://sourceforge.net/projects/samtools/files/samtools/0.1.13/samtools-0.1.13.tar.bz2 --no-check-certificate
$ tar -jxvf samtools-0.1.13.tar.bz2
$ cd /share/nas1/yuj/software/samtools-0.1.13
$ make
$ ./samtools
$ /share/nas1/yuj/software/samtools-0.1.13/samtools index /share/nas1/yuj/project/re-sequencing/GP-20230209-5591_20230317/analysis_dir/mapping_dir/01.bwa2sortbam/HHM.sort.bam &
Cite
"samtools index failed - 简书." https://www.jianshu.com/p/75ad536ef47b, 访问时间 1 Jan. 1970.
六.vcf
6.1 vcf拆分
python3 /share/nas1/yuj/script/ddradANDreseqANDgenome/vcf_tools/split_vcf_by_beds.py -v xxx.vcf -b bed_dir/ -o variants_vqsr_filter # 自带输出压缩
## 手动压缩
bgzip samples.snp.region.filter.vcf && tabix -p vcf samples.snp.region.filter.vcf.gz
6.2 vcf过滤
1.samples.gatk.con.snp.vqsr.filter.vcf
## 流程参数
vcftools --vcf $outdir/variation_dir/variants_anno_dir/samples.pop.$type[$i].anno.result.vcf --out $outdir/vcf_filter/samples.pop.$type[$i].filter \
--recode --minQ 30 \ # 保留质量值≥30的变异
--min-meanDP 5 \ # 平均测序深度≥5
--max-missing 0.70 \ # 允许最多30%的缺失数据
--remove-filtered-all \ # 移除所有被过滤的变异
--min-alleles 2 --max-alleles 2 # 仅保留双等位位点
&& perl $Bin/script/03vcf/format_filter.pl -i $outdir/vcf_filter/samples.pop.$type[$i].filter.recode.vcf -o $outdir/vcf_filter/samples.pop.$type[$i].filter.format.recode.vcf
## 自定参数
vcftools --gzvcf merged_final.sort.snp.vcf.gz \ # 输入压缩的VCF文件
--recode --recode-INFO-all --stdout \ # 保留所有INFO字段,输出到标准输出
--maf 0.05 \ # 保留次要等位基因频率≥5%的位点
--max-missing 1 \ # 不允许缺失数据(所有样本必须有基因型)
--minDP 10 \ # 单个样本的最小深度≥10
--maxDP 1000 \ # 单个样本的最大深度≤1000
--minQ 50 \ # 位点的最小质量值≥50
--minGQ 0 \ # 基因型质量无限制(GQ≥0)
--min-alleles 2 --max-alleles 2 \ # 仅保留二态位点(SNP)
--remove-indels \ # 移除所有Indel
--remove-filtered-all \ # 移除所有被过滤的位点
| gzip - > clean.vcf.gz # 压缩输出文件
## 更严格
vcftools --gzvcf merged_final.sort.snp.vcf.gz \
--recode --recode-INFO-all --stdout \
--maf 0.1 \
--max-missing 0.9 \
--minDP 20 \
--maxDP 1000 \
--minQ 100 \
--minGQ 20 \
--min-alleles 2 --max-alleles 2 \
--remove-indels \
--remove-filtered-all \
| gzip - > clean_strict.vcf.gz
2.对拆分后的vcf过滤
mkdir filter
ls *.con.snp.vqsr.filter.vcf.gz | perl -lpe 's/.con.snp.vqsr.filter.vcf.gz//' | parallel -j 100 "vcftools --gzvcf {}.con.snp.vqsr.filter.vcf.gz --recode --recode-INFO-all --maf 0.15 --max-missing 0.95 --minDP 50 --maxDP 1000 --minQ 100 --minGQ 50 --min-alleles 2 --max-alleles 2 --remove-indels --remove-filtered-all --out filter/{}.filter " # out为输出前缀
ls *.con.snp.vqsr.filter.vcf.gz | perl -lpe 's/.con.snp.vqsr.filter.vcf.gz//' | parallel -j 100 "vcftools --gzvcf {}.con.snp.vqsr.filter.vcf.gz --recode --recode-INFO-all --maf 0.05 --max-missing 0.9 --minDP 30 --maxDP 1000 --minQ 50 --minGQ 20 --min-alleles 2 --max-alleles 2 --remove-indels --remove-filtered-all --out filter/{}.filter "
6.3 vcf(或满足一二列要求的其他文件)合并
6.3.1 vcf相同样品合并不同区域(vcf并集)
cd filter
## for循环 排序
for f in samples.gatk.*.filter.recode.vcf; do bcftools sort $f -Oz -o sorted_${f}.gz && tabix sorted_${f}.gz & done
## 或者并行处理 排序
parallel -j 100 'bcftools sort {} -Oz -o sorted_{}.gz && tabix sorted_{}.gz' ::: samples.gatk.*.filter.recode.vcf
bcftools concat sorted_samples.gatk.*.filter.recode.vcf.gz -Oz -o merged.vcf.gz
6.3.2 其他文件相同样品合并不同区域(其他文件并集)
python3 /share/nas1/yuj/script/ddradANDreseqANDgenome/vcf_tools/merge_vcf-like-files.py -i *.file -o outfile
6.3.3 不同样品的vcf合并(vcf交集)
都是基于同样的参考基因组来的vcf文件
1.精确位点匹配(包括REF/ALT)
# 使用bcftools query提取变异标识符
bcftools query -f '%CHROM\t%POS\t%REF\t%ALT\n' sample1.vcf.gz | sort > sample1.sites
# 对其他样本做同样处理...
# 找出所有样本共有的位点
comm -12 sample1.sites sample2.sites | comm -12 - sample3.sites > common.sites
# 提取这些位点
bcftools view -T common.sites sample1.vcf.gz -Oz -o sample1_common.vcf.gz
# 对其他样本做同样处理...
# 最后合并
tabix sample1_common.vcf.gz
tabix sample2_common.vcf.gz
bcftools merge sample1_common.vcf.gz sample2_common.vcf.gz ... -o strict_merged_intersection.vcf
2.非严格匹配 bcftools isec + merge
# 首先找出所有样本共有的位点(交集)
bcftools isec -n=2 -w1 sample1.vcf.gz sample2.vcf.gz ... sampleN.vcf.gz -p dir_output
# 然后合并这些共有的位点
bcftools merge dir_output/0000.vcf dir_output/0001.vcf ... dir_output/000N.vcf -o merged_intersection.vcf
9️⃣.其他
9.1 主要分析步骤的整理(路径均以/share/nas1/yuj/pipline/reseq/v1.3为基准)
- 质量控制
sub fastQC {# 原始数据质控
# 使用ngsqc进行FastQC分析
$ngsqc = $config{"ngsqc"};
# 生成ATGC含量和测序质量分布图
$Rscript $Bin/script/00qc/ngsqc.R
- 基因组准备
sub IndexBuild {# 参考基因组索引构建
$bwa2 index $outdir/genome/$genome_name
# 生成GFF注释文件
grep -v '^#' $main->{Project}{Genome}{gff} > genome.gff
- 序列比对
# BWA比对和排序
$bwa2 mem | samtools sort
# 标记PCR重复
$gatk MarkDuplicates
- 比对统计
# 生成比对统计报告
$samtools stats
# 测序深度分析
$samtools depth
# 使用R绘制深度分布图
$Rscript depth_Track_point.r
- 变异检测(GATK流程)
# 生成gVCF文件
$gatk HaplotypeCaller --emit-ref-confidence GVCF
# 合并样本gVCF
$gatk CombineGVCFs
# 联合基因分型
$gatk GenotypeGVCFs
#
$gatk GatherVcfs
gatk SelectVariants
- 变异过滤
# VQSR过滤
$gatk VariantRecalibrator
$gatk ApplyVQSR
# 硬过滤
$gatk VariantFiltration
# 5.7
gatk Filter
- 注释分析
# SnpEff注释
$java -jar $snpEff
# 提取注释结果
extract_oneEff_anno.pl
# 生成SNP/INDEL列表
vcf_to_snplist_v1.4.pl
6.3 : gatk MergeVcfs
6.4 : gatk Result Stat
6.5 : Samples Diff Variation
- Step 7 : Vcftools Filter
1.过滤vcf
2.popSNP_Stat
# 变异统计报表
$bcftools stats
# 转换类型统计(Ti/Tv)
titv_stat.py
# 变异长度分布统计
length_indel.pl
- 结构变异检测
sub sv{
# 使用lumpyexpress检测SV
$lumpyexpress -B ... -S ... -D ...
# SV基因型判定
$svtyper
# SV注释
$snpEff
}
- 09 : Variant Gene Annotation
----报告内容
变异在基因组上的分布--circos软件展示
## 3.7 BSA关联分析
### 3.7.3 关联区域内基因注释
GO
COG
通路代谢
KEGG
KEGG富集分析--散点图
----对应软件
数据质控--fastp
比对参考基因组--bwa 输出sam文件
转换--samtools 输出bam文件
gatk_haplotypecaller--变异位点
gatk_VariantFiltration--过滤
snpeff--注释变异(SNP、Small InDel)和预测变异影响
breakdance--sv结构变异检测
vcftools--获取可信变异位点
python脚本&ggplot2--混池SNPindex数据拟合&绘图
blastall--区间内基因功能注释
primer3--设计区间内indel标记的引物
----分析流程
step7 过滤后获取可信变异位点
step8 sv结构变异检测
step9 区间内基因注释