05线粒体流程3-高级分析
1.参考基因组数据准备
1.1下载
## 复制表格中的物种 至 tmp_adv文件(以+号为分割)
编号目录]$ awk -F "+" '{ print $2 }' tmp_adv | awk '$1=$1' > list_adv && cat list_adv && down- adv && cd ref_adv/ && cat fasta/* > allref.fa && cd ../
## 把所有组装结果复制过来,受后缀名影响
for i in $( ls analysis/assembly) ;do echo $i;cp analysis/assembly/$i/finish/*.fsa ref_adv/$i.fsa ;done
for i in $( ls analysis/assembly) ;do echo $i;cp analysis/annotation/$i/$i.gbk ref_adv/;done
1.2改名
## 利用awk提取一下名字
# cd ref_adv/fasta && for i in *.fasta;do awk '{if(NR==1) print $0}' $i;done | awk -F ">" '{ print $2 }' | awk -F ".1_" '{ print $2"""_"""$1""".1" }' && cd -
# print a""b >>> 结果 ab
1.添加一下登录号信息
cd ref_adv
mv gbk gbk2 && mkdir gbk && cd gbk2 && for i in *.gbk;do num=`awk 'NR==1 {print $2}' $i` && echo "sed -e '/ORGANISM/s/$/ "$num"/' -e '/\/organism=/s/\"$/ "$num"\"/' "$i" > ../gbk/"$i >> modified1.sh;done && sh modified1.sh && cd ../../
2.给gbk、fasta改名
GP-XXX]$ cd ref_adv/gbk && rename .gbk .1.gbk * && cd - # 加上.1,一般不需要
GP-XXX]$ cd ref_adv/fasta && rename .fasta .1.fasta * && cd - # 加上.1,一般不需要
cd ref_adv/fasta && for i in *.fasta;do rename `awk '{if(NR==1) print $0}' $i | awk -F ">" '{ print $2 }' | awk -F ".1_" '{ print ""$1""".1" }'` `awk '{if(NR==1) print $0}' $i | awk -F ">" '{ print $2 }' | awk -F ".1_" '{ print $2"""_"""$1""".1" }'` ../gbk/""`awk '{if(NR==1) print $0}' $i | awk -F ">" '{ print $2 }' | awk -F ".1_" '{ print ""$1""".1" }'`.gbk ;done && cd ../../
cd ref_adv/fasta && for i in *.fasta;do rename `awk '{if(NR==1) print $0}' $i | awk -F ">" '{ print $2 }' | awk -F ".1_" '{ print ""$1""".1" }'` `awk '{if(NR==1) print $0}' $i | awk -F ">" '{ print $2 }' | awk -F ".1_" '{ print $2"""_"""$1""".1" }'` ../fasta/""`awk '{if(NR==1) print $0}' $i | awk -F ">" '{ print $2 }' | awk -F ".1_" '{ print ""$1""".1" }'`.fasta ;done && cd ../../
1.3检查
cd ref_adv
mt_get_cds.py -i gbk/ -o out_all -d
1.4矫正(考虑全基因组水平的pi分析)
共线性好的话,修改起点
cd ref_adv/fasta
for i in *.fasta*;do nuc ../*.fsa $i -l 1 && awk 'NR==4 {print $3}' out.delta ;done
for i in *.fasta*;do nuc ../*.fsa $i -l 1 && mt_move_pos.py -fa $i -s `awk 'NR==4 {print $3}' out.delta` ;done
cd ../ && mkdir pi && mv fasta/*.fsa2 pi && cd pi && rename fsa2 fasta *.fsa2 && cd ../ && cat pi/*.fasta *.fsa > pi.fa
nuc *.fsa pi.fa && mum
二.高级分析
1.获取高级分析配置
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/advance/advanced.yaml.final.pl -i analysis/ -f ref_adv/fasta/ -g ref_adv/gbk/
-tree ref_tree/all.fa
# 建议把高级分析物种按物种名排序,也就是改文件名
!!!务必注意,组装样品gbk的位置是否有多余gbk!!!
2.主流程
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/advance/advanced_pip.v1.pl -i advanced.config.yaml -g 2/5
# 跑流程 2是脊椎动物 5是无脊椎动物
三.检查结果
3.1 cgview圈图
#对应该步命令,但该程序会生成final.xml文件,覆盖掉修改的参数,如高宽参数
cd analysis]
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/current/cgview_comparison/cgview_cmp_gbk.pl -i ../ref_adv/sample/gbk/Mm_G1.gbk -c ../ref_adv/gbk/*.gbk -o cgview &
1.若有重叠
cd analysis/cgview
## vi maps/cgview_xml/mtDNAsmall.xml
## height="1050" width="1050" 修改高宽参数
sed -i 's/height="1000"/height="1050"/g' maps/cgview_xml/mtDNAsmall.xml
sed -i 's/width="1000"/width="1050"/g' maps/cgview_xml/mtDNAsmall.xml
java -jar -Xmx1500m /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v1.0/cgview_comparison/etc/cgview_comparison_tool/bin/cgview.jar -i maps/cgview_xml/mtDNAsmall.xml -o mtDNA.cgview_cmp.svg -f svg
svg2xxx -t png -dpi 300 mtDNA.cgview_cmp.svg && svg2xxx -t pdf mtDNA.cgview_cmp.svg
3.2 kaks选择压力分析
3.2.1 程序
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/advance/kaks/kaks.each.two.pl -i 样品.gbk -r ref们.gbk -o 输出 -g 密码子表
cd kaks && ll */*xls
3.2.2 多物种kaks比较画图
1.取出流程里的所有表格并合并
!!!用"_"做分隔符,要单独考虑“NC_”,所以这一步分成了两个文件夹 !!!
样品编号]
mkdir kaks_plot/nc -p && mkdir kaks_plot/other -p
cp analysis/kaks/*/*.xls kaks_plot && cd kaks_plot && mv *NC*.xls nc ; mv *.xls other
2.不同项目文件名不一样,"_"的个数也不一样,到时候好好数数把最后一段提取出来
cd other && for i in *.xls;do tail -n +8 $i | awk -F "\t" '{print $1 "\t" $5}' | awk -F "_" '{print $4}' > $i.txt;done
cd ../nc && for i in *.xls;do tail -n +8 $i | awk -F "\t" '{print $1 "\t" $5}' | awk -F "_" '{print $5}' > $i.txt;done
先看下是否提取对了
R 箱线图1
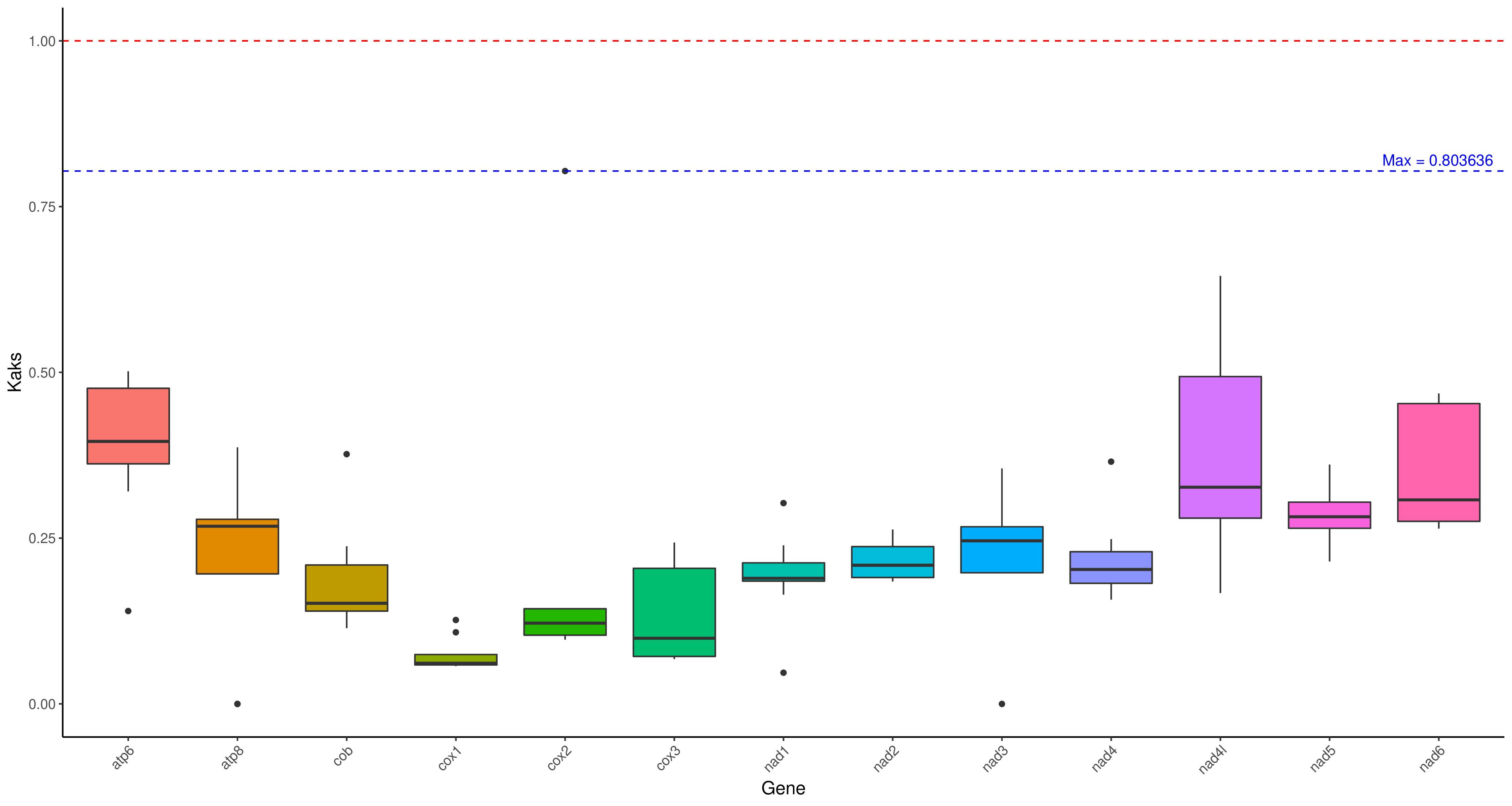
cd ../ && mv */* ./
python3 /share/nas1/yuj/script/chloroplast/advance/kaks/merged.py -2 # 在这个目录里,就这么运行就行,会生成merged.txt
## 将NA替换为0
sed 's/NA/0/g' merged.txt > gene.kaks.xls
Rscript /share/nas1/yuj/program/chloroplast/kaks/boxplot_kaks.R gene.kaks.xls gene.kaks.pdf && convert -density 300 gene.kaks.pdf gene.kaks.png
R 箱线图2
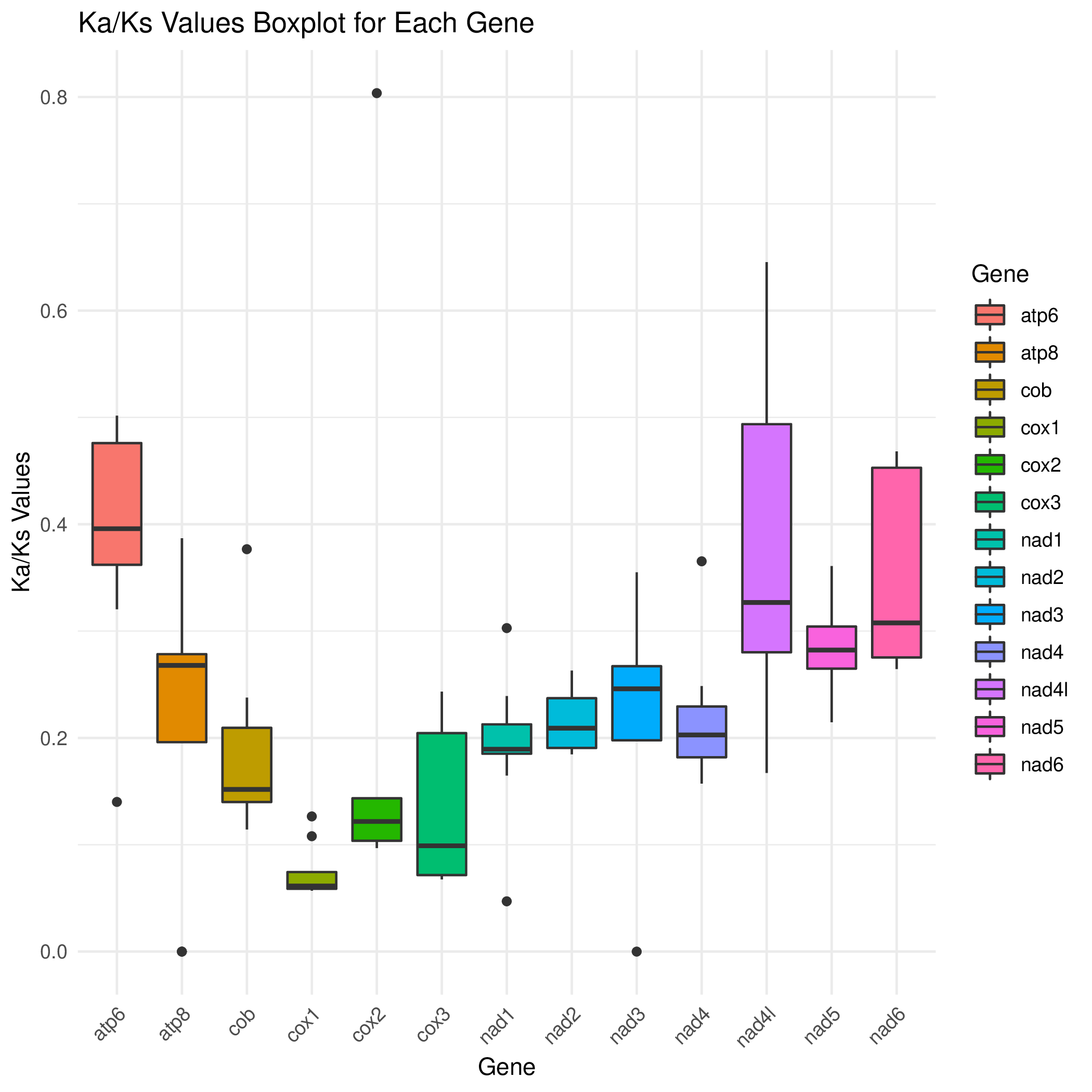
cd ../ && mv */* ./
python3 /share/nas1/yuj/script/chloroplast/advance/kaks/merged.py -1 # 在这个目录里,就这么运行就行,会生成merged.txt
## 将NA替换为0
sed -i 's/NA/0/g' merged.txt # 用命令吧
head_info=`files=$(ls *.xls) && echo -e "${files}" | sed 's/\.all\.gene\.kaks\.stat\.xls//g' | tr '\n' '\t' | sed 's/\t$//'`
cp merged.txt genekaks.txt
sed -i "1i Gene\t$head_info" genekaks.txt
cp /share/nas1/yuj/script/chloroplast/advance/kaks/boxplot_kaks.R ./
Rscript boxplot_kaks.R && convert -density 300 gene.kaks.pdf gene.kaks.png
柱状图
绘图数据获取,同python箱线图
cp /share/nas1/yuj/script/chloroplast/advance/kaks/kaks_plot.py ./
手动修改程序里的参数再运行
python3 kaks_plot.py
3.2.3 atp8未计算
原因是两两比对的E值大于/share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/advance/kaks/src/kaks.two.pl中blast解析程序设置的evalue值
建议更改/share/nas1/yuj/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/advance/kaks/src/kaks.two.pl程序
第120行,加上 -eval xxx
3.3 同源性比对
Mauve
Mauve & # 用第二种比对方式
mVISTA
1.网站
[VISTA tools](http://genome.lbl.gov/vista/index.shtml)
2.转换注释
perl /share/nas1/yuj/script/chloroplast/advance/get_mVISTA_format_from_GenBank_annotation.pl -i gbk -p .gbk # -i输入文件夹 -p输入文件后缀
3.上传所有物种fasta、参考物种注释
把参考物种上传两遍,因为第一个参考作参考后不显示该基因组
4.选择Shuffle-LAGAN比对
5.打开结果页面,选第一个物种,点击"VISTA-Point"
6.结果页面,点击第一个物种右侧pdf下载
3.4 phytree
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/advance/phytree/raxml_phytree.pl
3.5 全局比对(snp indel) 一般不做
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/advance/global_align/global_align.pl
# 同叶绿体
perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/script/global_align/global_align.pl -i 组装出的基因组(一般是第一个) -a 一般是第一个物种注释 -r 参考基因组/其他物种.fa -o global_align
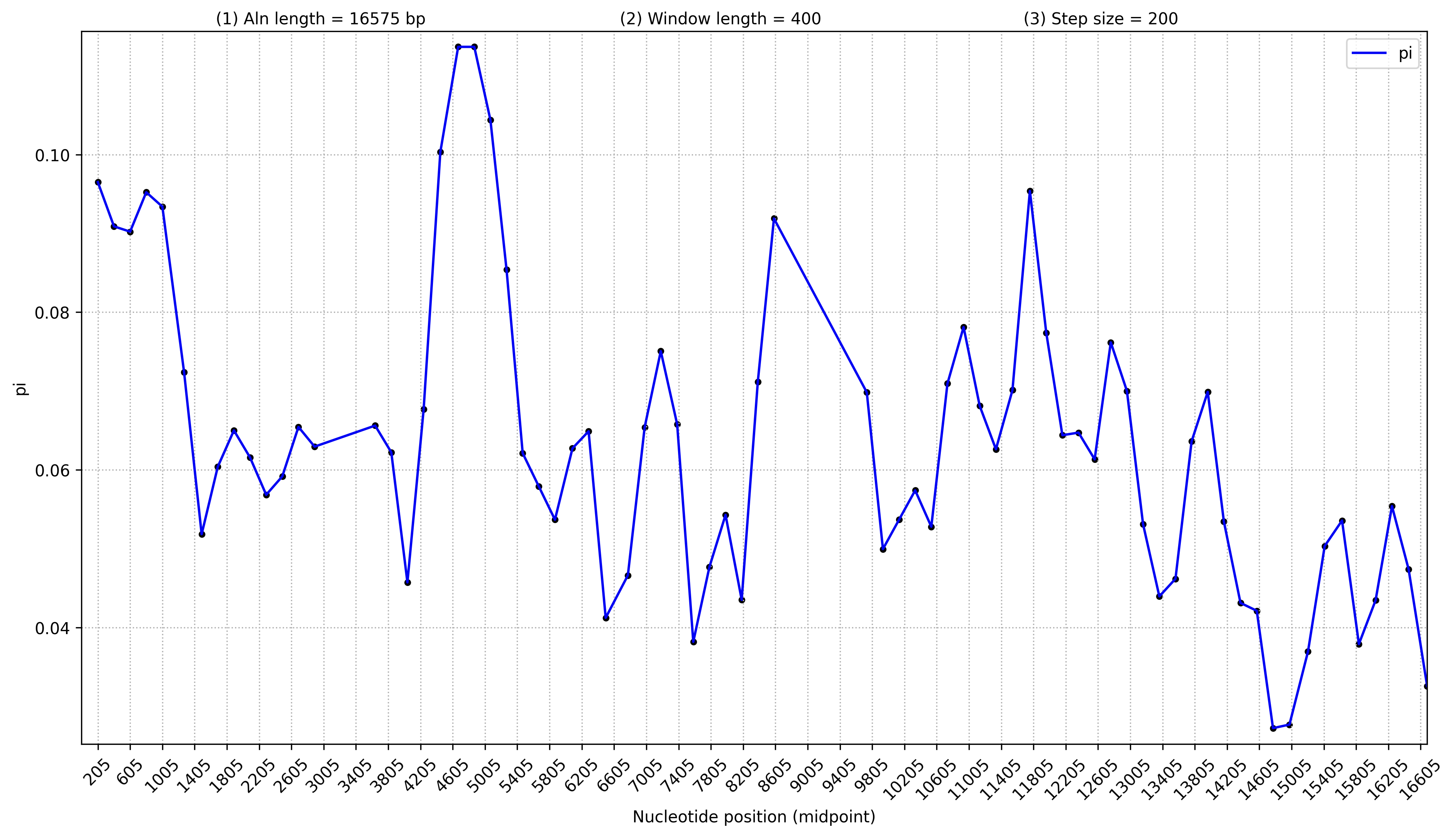
3.6 核酸多样性分析(默认不分析,分析的话先完成1.4)
不显示基因
mafft --auto --quiet --thread 30 allinone.fa > allinone.aln # 有点费时
perl /share/nas6/xul/program/mt2/phytree/gene_tree/src/fasta2line.pl -i allinone.aln -o all.fasta.line
python3 /share/nas1/yuj/script/chloroplast/advance/pi/full_genome_calculate_pi.py all.fasta.line 400 200 > pi.stat.xls
python3 /share/nas1/yuj/script/chloroplast/advance/pi/mt_full_genome_pi_plot.py -i pi.stat.xls -o pi.full.png # 4节点运行会报错,cluster节点不会报错
显示基因
1.比对
mafft --auto --quiet --thread 30 allinone.fa > allinone.aln # 有点费时
perl /share/nas6/xul/program/mt2/phytree/gene_tree/src/fasta2line.pl -i allinone.aln -o all.fasta.line
2.滑窗计算
python3 /share/nas1/yuj/script/chloroplast/advance/pi/full_genome_calculate_pi.py all.fasta.line 200 20 > pi.stat.xls
3.获取比对后基因位置
python3 /share/nas1/yuj/script/chloroplast/advance/pi/position_mapping.py -i all.fasta.line -p Ganoderma_sinense -i2 *.gbk.ann -o2 gene.range.info
4.绘图
mamba activate pylatest
python3 /share/nas1/yuj/script/chloroplast/advance/pi/mt_full_gene_pi_plot.py -i1 pi.stat.xls -i2 gene.range.info -o pi.gene.png
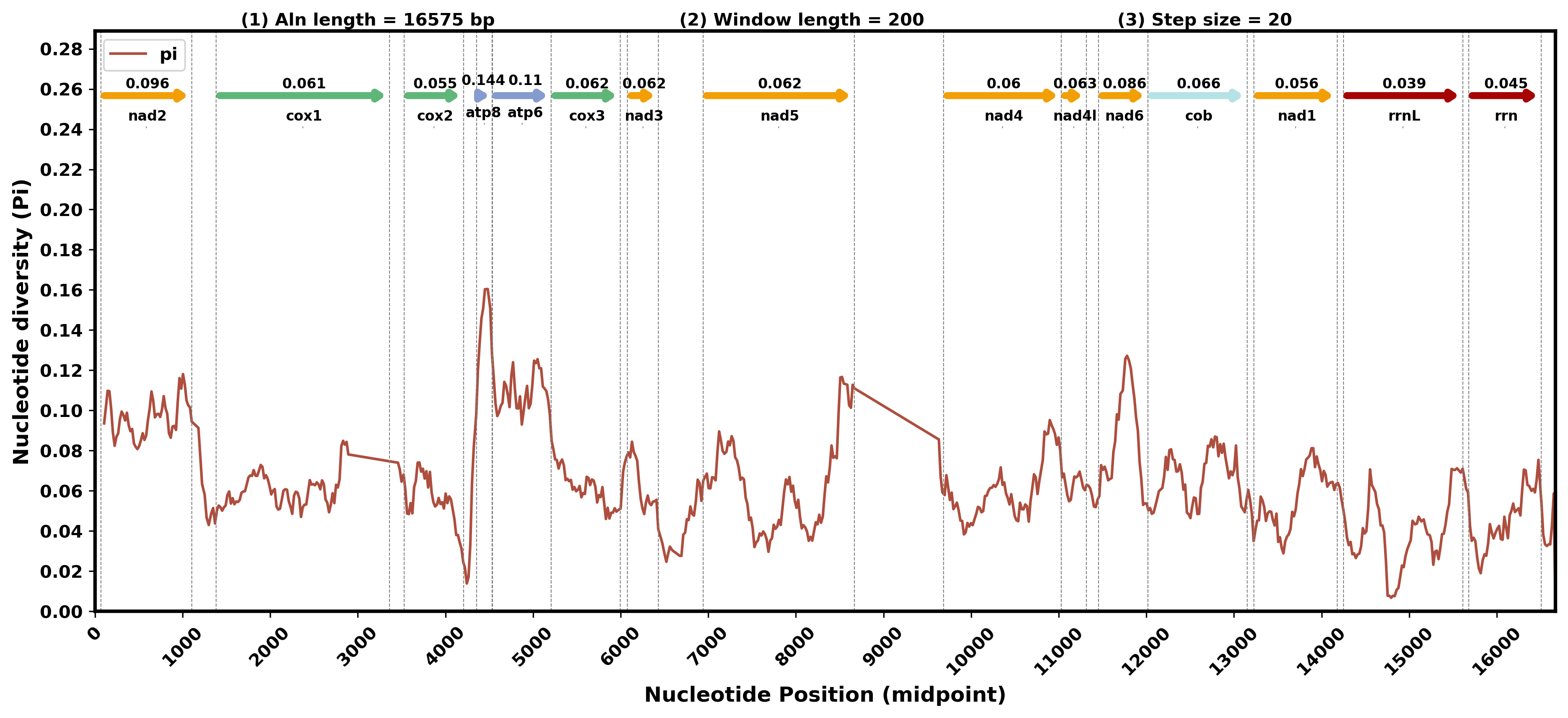
四.整理结果
## 检查图片
mkdir fig && cp analysis/annotation/*/ogdraw/*.dpi300.circular.png analysis/annotation/*/asmqc/coverage_plot/*.png analysis/annotation/*/asmqc/cmp.dotplot.png fig
## 整理
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/html_report/mt_pip_dir.pl -i analysis
rm -rf complete_dir/03Asmqc/*/*.coverage.svg
---- 华大数据 ----
perl /share/nas1/yuj/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/annotation/src/fastq_qc_wgs_bgi.pl -i analysis/samples.reads.txt -o complete_dir/01Rawdata
0.原始数据统计 可能出问题
/share/nas6/zhouxy/modules/ngsqc/ngsqc_bgi -1 fq1 -2 fq2 -k 物种名 -o ./(华大可用.)
/share/nas6/zhouxy/biosoft/bin/ngsqc -1 fq1 -2 fq2 -k 物种名 -o ./ (华大不能用)
Rscript /share/nas6/xul/program/reseq/v1.1/script/ngsqc.R --base *.atgc --qual *.qual --out ./
sub qc{
system "/share/nas6/zhouxy/biosoft/bin/ngsqc -1 $_[1] -2 $_[2] -k $_[0] -o . ";
system "Rscript /share/nas6/xul/program/reseq/v1.1/script/ngsqc.R --base $_[0].atgc --qual $_[0].qual --out $_[0]";
}
for my $svg (@svg){
system "svg2xxx -t pdf $svg.acgtn.svg";
system "svg2xxx -t png -dpi 600 $svg.acgtn.svg";
system "svg2xxx -t pdf $svg.quality.svg";
system "svg2xxx -t png -dpi 600 $svg.quality.svg";
五.生成报告
图片路径 /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/html_report/src/tmp
1.修改报告配置文件
cp /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/html_report/report.cfg ./report.cfg && realpath report.cfg
for i in `cat ann.cfg |grep gbk | cut -f 4`;do echo "#####################"$i && head $i;done
2.标准分析
## 标准分析,不包含测序
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/html_report/report2xml.xianliti2.pl -id complete_dir/ -cfg report.cfg
## 标准分析 后面有-n
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/html_report/report2xml.xianliti.pl -id complete_dir/ -cfg report.cfg -n
3.标准+高级分析,标准分析用也行
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/html_report/report2xml.xianliti.pl -id complete_dir/ -cfg report.cfg
4. 三代辅助组装
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/html_report/report2xml.xianliti.3dai.pl -id complete_dir/ -cfg report.cfg
PS:先单独生成3代统计
perl /share/nas6/xul/program/mt2/tgs_reads_statistic/multi_tgs_qc.pl -i analysis/samples.reads.txt -o complete_dir/01Rawdata/
其中samples.reads.txt第四列是3代路径
5.容易出问题的质控
perl /share/nas6/pub/pipline/genome-assembly-seq/mitochondrial-genome-seq/v2.0/asmqc/mtDNA_asmqc.pl -i 组装结果 -1 clean*1 -2 clean*2 -p 物种名 -q 物种.gbk -o asmqc -g 2 -r 参考.gbk