04叶绿体流程3-高级分析
1.参考基因组数据准备
1.1 下载ref
## 复制表格中的物种 至 tmp_adv文件(以+号为分割)
编号目录]
awk -F "+" '{ print $2 }' tmp_adv | awk '$1=$1' > list_adv && cat list_adv && down- adv && cd ref_adv/ && cat fasta/* > allref.fa && cd ../
或
python3 /share/nas1/yuj/script/chloroplast/download_genome_from_ncbi.py -f gb -a list_adv # 备用下载
## 把所有组装结果复制过来,受后缀名影响
for i in $( ls analysis/assembly) ;do echo $i;cp analysis/assembly/$i/finish/*.fsa ref_adv/$i.fsa ;done
for i in $( ls analysis/assembly) ;do echo $i;cp analysis/annotation/$i/gene_anno/$i.gbk ref_adv/;done
1.2矫正
1.查看长度 去掉长度明显异常的(如4204项目的高级分析物种掺入了线粒体)
GP-XXX]$ cd ref_adv/fasta && for i in * ;do ir.py -i $i -l;done && cd ../../
2.看nuc程序结果 判断各物种序列情况(ssc反向/整体反向)
$ cd ref_adv && cat fasta/* *.fsa > all.fa && nuc *.fsa all.fa && mum
有问题的转至步骤4注释,其他正常的继续按步骤查看
$ mv xxx ../ && mkdir ann
$ ir.py -i xxx -o xxx.fasta2 # 整体反向调整,获得正确的序列
$ mt_move_pos.py -fa xxx.fasta2 -s 10086
$ cp_ssc.pl -i xxx -o ann/xxx.fasta # ssc反向调整,获得正确的序列
3.看ir程序结果 判断各物种序列情况(是否起点有错)
$ cd fasta/ && for i in *.fasta;do ir $i ;done > ir.log && cd ../ && cat fasta/ir.log
# (单个修改)起点不对,就执行下面两条命令修改 .gbk/.fasta文件
# ref_adv]$ perl /share/nas6/xul/program/chloroplast/bin/cp_format_gbk.pl -f gbk/NC_054249.1.gbk -s 14
# for i in new_gbk/*.gbk;do echo $i;/share/nas6/xul/program/chloroplast/bin/cp_gbk2fasta.pl -i $i -o new_gbk;done
(1) (批量修改)起点不对
ref_adv]
python3 /share/nas1/yuj/script/chloroplast/phytree/cp_batch_adjust_genome_start.py -i1 fasta/ -i2 gbk/
停顿几秒等运行结束
for i in new_gbk/*.gbk;do echo $i;/share/nas6/xul/program/chloroplast/bin/cp_gbk2fasta.pl -i $i -o new_gbk;done && rm complete2 -r
(2) 执行下面命令再检查一遍
ref_adv]$ cd new_gbk/ && for i in *.fasta;do rename "_no_IR" "" $i;done && for i in *.gbk;do mv $i ${i%_no_IR.gbk}.gbk;done && for i in *;do ir $i;done && cd ../
# 可能会出现ir区不是完全相似的情况,但不影响这一步的检查目的
(3) 第(2)步没问题的话,这一步替换原文件夹里的文件
ref_adv]$ \mv -f new_gbk/*.fasta fasta/ && \mv -f new_gbk/*.gbk gbk/ && rm new_gbk -r
# cd ref_adv/fasta/ && for i in *;do ir $i;done && cd ../../
# cd ref_adv/gbk/ && for i in *;do ir $i;done && cd ../../
4.对参考进行重新注释
(1)具体步骤
cd ref_adv/ann
for i in *.fasta;do echo $i;mkdir -p analysis/assembly/${i%.*}/finish;done
for i in *.fasta;do echo $i;mv $i analysis/assembly/${i%.*}/finish && rename .fasta _FULLCP.fsa analysis/assembly/${i%.*}/finish/$i;done
python3 /share/nas1/yuj/script/chloroplast/get_ann_cfg.py
填上gbk路径
perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/annotation/bin/chloroplast_annotaion.pl.v2.1.pl -d analysis/assembly/ -i ann.cfg
# 修改注释
cd ref_adv
python3 /share/nas1/yuj/script/chloroplast/phytree/cp_from_gbk_get_cds_V4.0.py -i gbk/ -o out/ # 提取cds和trna
把需要修改的物种放在archive中
cd fasta/archive && for i in MZ*.fasta;do echo $i;cp ../../cds/"trna_"$i ../analysis/annotation/${i%.1*}/gene_anno;done && for i in MZ*.fasta;do echo $i;cp ../../cds/"cds_"$i ../analysis/annotation/${i%.1*}/gene_anno;done
具体某个物种]$
# 显示trnH-GUG的序列
$ sed -n "$(let a=`awk '/trnH-GUG/{print NR}' trna_*.fasta`+1 && echo $a) p" trna_*.fasta > trnh
$ blastn -query trnh -subject *.fsa -outfmt 6
# 显示trnS-UGA的序列
$ sed -n "$(let a=`awk '/trnS-UGA/{print NR}' trna_*.fasta`+1 && echo $a) p" trna_*.fasta > trns
$ blastn -query trns -subject *.fsa -outfmt 6
# 显示petD的序列
$ sed -n "$(let a=`awk '/petD/{print NR}' cds_*.fasta`+1 && echo $a) p" cds_*.fasta > petD
$ blastn -query petD -subject *.fsa -outfmt 6
(2)若上面做错了,则重新复制到finish
ann]$ for i in $( ls analysis/assembly) ;do echo $i;cp analysis/assembly/$i/finish/* analysis/$i.fasta ;done # 不受后缀名影响
# 然后重复上面(1)步骤
(3)注释完复制过去
cd ann
for i in `ls analysis/assembly/`;do cp analysis/assembly/$i/finish/*.fsa ../fasta/$i.fasta; cp analysis/annotation/$i/gene_anno/$i.gbk ../gbk/;done
1.3高级分析配置文件按物种名排序gbk,不依据登录号
## 利用awk提取一下名字
# cd ref_adv/fasta && for i in *.fasta;do awk '{if(NR==1) print $0}' $i;done | awk -F ">" '{ print $2 }' | awk -F ".1_" '{ print $2"""_"""$1""".1" }' && cd -
# print a""b >>> 结果 ab
1.添加一下登录号信息
## 直接用这个程序修改
!!!!!!!!!!!!!!!!!!!
python3 /share/nas1/yuj/script/chloroplast/advance/cp_irscope.py -i ref_adv/ -s ycf1
## 下面不用了
# cd ref_adv
# mv gbk gbk2 && mkdir gbk && cd gbk2 && for i in *.gbk;do num=`awk 'NR==1 {print $2}' $i` && echo "sed -e '/ORGANISM/s/$/ "$num"/' -e '/\/organism=/s/\"$/ "$num"\"/' "$i" > ../gbk/"$i >> modified1.sh;done && sh modified1.sh && cd ../../
2.给gbk、fasta改名
GP-XXX]$
cd ref_adv/gbk && rename .gbk .1.gbk * && cd - # 加上.1,一般不需要
cd ref_adv/fasta && rename .fasta .1.fasta * && cd - # 加上.1,一般不需要
cd ref_adv/fasta && for i in *.fasta;do rename `awk '{if(NR==1) print $0}' $i | awk -F ">" '{ print $2 }' | awk -F ".1_" '{ print ""$1""".1" }'` `awk '{if(NR==1) print $0}' $i | awk -F ">" '{ print $2 }' | awk -F ".1_" '{ print $2"""_"""$1""".1" }'` ../gbk/""`awk '{if(NR==1) print $0}' $i | awk -F ">" '{ print $2 }' | awk -F ".1_" '{ print ""$1""".1" }'`.gbk ;done && cd ../../
cd ref_adv/fasta && for i in *.fasta;do rename `awk '{if(NR==1) print $0}' $i | awk -F ">" '{ print $2 }' | awk -F ".1_" '{ print ""$1""".1" }'` `awk '{if(NR==1) print $0}' $i | awk -F ">" '{ print $2 }' | awk -F ".1_" '{ print $2"""_"""$1""".1" }'` ../fasta/""`awk '{if(NR==1) print $0}' $i | awk -F ">" '{ print $2 }' | awk -F ".1_" '{ print ""$1""".1" }'`.fasta ;done && cd ../../
二.高级分析
1.获取高级分析配置
perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/script/advanced.yaml.final.pl -i analysis/ -f ref_adv/fasta -g ref_adv/gbk
# 有默认输出,不用改(如果前面步骤没出错)
2.主流程
perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/script/advanced.analysis/advanced_pip.v1.pl -i analysis/advanced.config.yaml -o analysis/ &
三.检查结果
3.1Cgview圈图(20230313已去掉/202304加回来)
/share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/cgview_comparison/cgview_cmp_gbk.pl
# 对应该程序,但该程序会生成final.xml文件,覆盖掉修改的参数,如高宽参数
/share/nas1/yuj/project/chloroplast/GP-20230131-5554_20230227/analysis/commands_dir/cgview.commands
# 对应该命令
1.根据物种名排列作图,需要把命令里对应的文件名字改了,文件名改,命令里输入的顺序也要改
analysis/cgview]$ perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/cgview_comparison/cgview_cmp_gbk.pl -i 物种.gbk -c .../2.1.gbk .../3.1.gbk -p 物种名 -o ./
2.查看图片,若有重叠
cd analysis/cgview
## vi maps/cgview_xml/cpDNAsmall.final.xml
## height="1050" width="1050" 修改高宽参数
sed -i 's/height="1000"/height="1050"/g' maps/cgview_xml/cpDNAsmall.final.xml
sed -i 's/width="1000"/width="1050"/g' maps/cgview_xml/cpDNAsmall.final.xml
java -jar -Xmx1500m /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/cgview_comparison/etc/cgview_comparison_tool/bin/cgview.jar -i maps/cgview_xml/cpDNAsmall.final.xml -o cpDNA.cgview_cmp.svg -f svg # 重新绘图
svg2xxx -t png -dpi 600 cpDNA.cgview_cmp.svg && svg2xxx -t pdf cpDNA.cgview_cmp.svg #cpDNA.cgview_cmp.svg是已有文件
# PS: 最终图里的名字 只与文件名关联
3.2 Kaks选择压力分析
3.2.1 检查结果
# 展示出所有.xls文件大小
irscope]$ cd ../kaks/ && ll */*xls
3.2.2 多物种绘制(植物线粒体程序)
cd analysis/kaks
Rscript /share/nas1/yuj/program/chloroplast/kaks/boxplot_kaks.R gene.kaks.xls gene.kaks.pdf && convert -density 300 gene.kaks.pdf gene.kaks.png
3.2.3 自定义作图
1.取出流程里的所有表格并合并
!!!用"_"做分隔符,要单独考虑“NC_”,所以这一步分成了两个文件夹 !!!
样品编号]
mkdir kaks_plot/nc -p && mkdir kaks_plot/other -p
cp analysis/kaks/*/*.xls kaks_plot && cd kaks_plot && mv *NC*.xls nc ; mv *.xls other
2.不同项目文件名不一样,"_"的个数也不一样,到时候好好数数把最后一段提取出来
cd other && for i in *.xls;do tail -n +8 $i | awk -F "\t" '{print $1 "\t" $5}' | awk -F "_" '{print $4}' > $i.txt;done
cd ../nc && for i in *.xls;do tail -n +8 $i | awk -F "\t" '{print $1 "\t" $5}' | awk -F "_" '{print $5}' > $i.txt;done
R 箱线图1
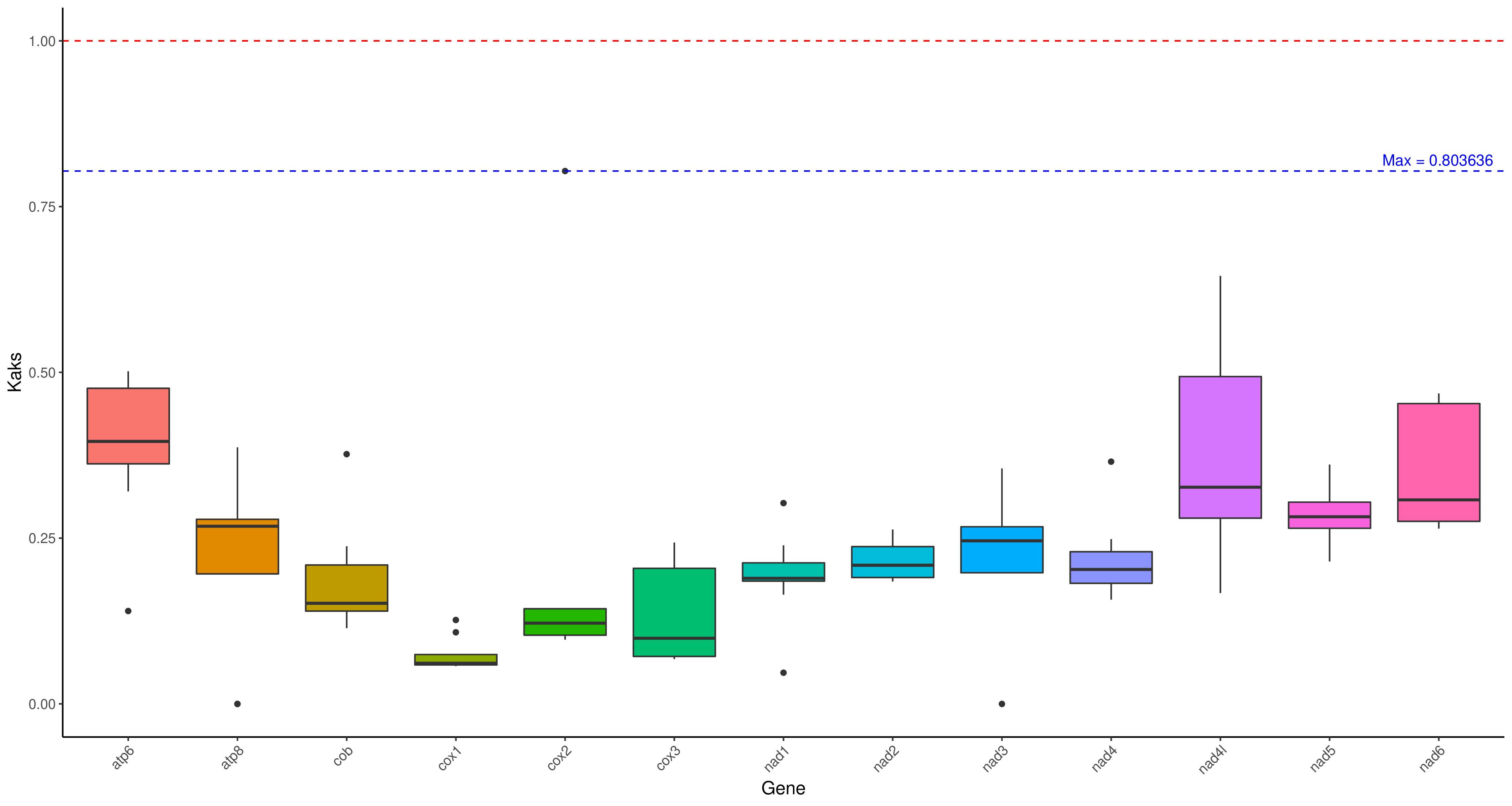
cd ../ && mv */* ./
python3 /share/nas1/yuj/script/chloroplast/advance/kaks/merged.py -2 # 在这个目录里,就这么运行就行,会生成merged.txt
## 将NA替换为0
sed 's/NA/0/g' merged.txt > gene.kaks.xls
Rscript /share/nas1/yuj/program/chloroplast/kaks/boxplot_kaks.R gene.kaks.xls gene.kaks.pdf && convert -density 300 gene.kaks.pdf gene.kaks.png
R 箱线图2
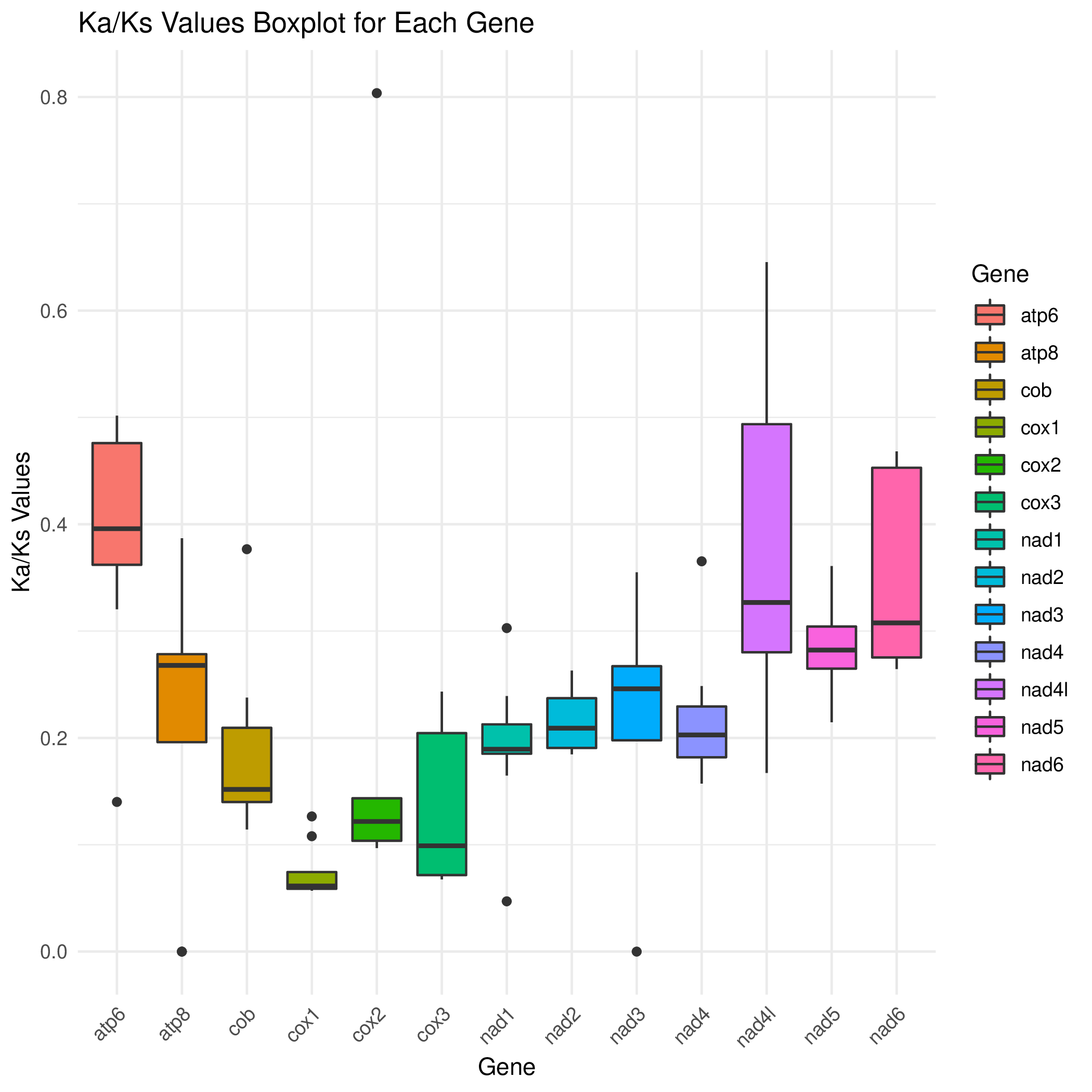
cd ../ && mv */* ./
python3 /share/nas1/yuj/script/chloroplast/advance/kaks/merged.py -1 # 在这个目录里,就这么运行就行,会生成merged.txt
## 将NA替换为0
sed -i 's/NA/0/g' merged.txt # 用命令吧
head_info=`files=$(ls *.xls) && echo -e "${files}" | sed 's/\.all\.gene\.kaks\.stat\.xls//g' | tr '\n' '\t' | sed 's/\t$//'`
cp merged.txt genekaks.txt
sed -i "1i Gene\t$head_info" genekaks.txt
cp /share/nas1/yuj/script/chloroplast/advance/kaks/boxplot_kaks.R ./
Rscript boxplot_kaks.R && convert -density 300 gene.kaks.pdf gene.kaks.png
柱状图
绘图数据获取,同python箱线图
cp /share/nas1/yuj/script/chloroplast/advance/kaks/kaks_plot.py ./
手动修改程序里的参数再运行
python3 kaks_plot.py
散点图
python3 /share/nas1/yuj/script/chloroplast/advance/kaks/scatter_plot_kaks_values.py 1Navicula_incerta_CACC_0356.kaks.xls gene.kaks1.png
输入文件$sample.kaks.xls
箱线图2里面的genekaks.txt就是
Gene KY499654 NC_015403 NC_044464 NC_056790 NC_056794
atpA 0.077968 0.0629167 0.0694429 0.0603908 0.0707976
atpB 0.0659463 0.07844 0.0725418 0.0737089 0.0585994
atpD 0.171715 0.154146 0.235884 0.16049 0.178153
atpE 0.0767651 0.029999 0.0288074 0.032989 0.0432521
atpF 0.194199 0.196788 0.188477 0.198927 0.116758
3.3 PI核酸多样性分析
3.3.1 CDS (流程图)
## 完整分析对应以下程序
perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/script/pi.analysis/pi.analysis.pl
-i /share/nas1/yuj/project/GP-20220331-4159-1_20220509/analysis/pi/gbk
-ir 104009-119152,138824-153967
-p Huperzia_crispata -o 0818 # -p必须是测序样品名
## 仅绘图
perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/script/pi.analysis/src/plot.pi.pl -i pi.stat.xls -o pi.gene.svg
perl /share/nas1/yuj/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/script/pi.analysis/src/plot.pi.pl -i pi.stat.xls -o pi.gene.svg
# 去掉旋转 20240925按客户要求,y轴字体12,x轴字体11
## 检查
kaks]$ cd ../pi && cat pi.stat.xls | sort -k 3 -n
&& display pi.gene.png
pi]$ display pi.gene.png #看值的大小不超过10,百分之10
pi]$ cat pi.stat.xls
pi]$ cat pi.stat.xls | cut -f 3
pi]$ cat pi.stat.xls | sort -k 3
pi]$ cat pi.stat.xls | sort -k 3 -n #挑第三列来排序,看第三列最后一个值大小
pi]$ ll mafft/ # 有问题的话找这
| 标记点 | 长度 (bp) | 多态位点 (个) | 简约信息位点 (个) | 平均遗传距离 | 核苷酸多样性 |
|---|---|---|---|---|---|
| rbcL-accD | 3283 | 34 | 3 | 0.01924 | 0.01877 |
| ycf1a | 1073 | 57 | 27 | 0.02407 | 0.02440 |
| ycf1b | 1860 | 108 | 31 | 0.02861 | 0.02881 |
| trnH-pabA | 434 | 12 | 2 | 0.01031 | 0.00989 |
| rbcL | 1434 | 12 | 2 | 0.00311 | 0.00311 |
| matK | 1521 | 20 | 4 | 0.00491 | 0.00491 |
3.3.2 Full Genome (非流程)
1.没有基因名、四分区域
mafft --auto --quiet --thread 30 allinone.fa > allinone.aln # 有点费时
perl /share/nas6/xul/program/mt2/phytree/gene_tree/src/fasta2line.pl -i allinone.aln -o all.fasta.line
python3 /share/nas1/yuj/script/chloroplast/advance/pi/full_genome_calculate_pi.py all.fasta.line 600 50 > pi.stat.xls # 800 100看起来差不多
mamba activate pylatest
python3 /share/nas1/yuj/script/chloroplast/advance/pi/cp_full_genome_pi_plot.py -i pi.stat.xls -o pi.full.png # 4节点运行会报错,cluster节点不会报错
mamba deactivate
获得的图片没有基因名
2.区域名(-i 输入文件最后一列是区域)
http://112.86.217.82:9919/#/tool/alltool/detail/328
上传allinone.fa或all.fasta.line,上传样品gbk文件,参数400,200
获取结果pi.stat.xls、gap_pos_for_real_pos.xls
mamba activate pylatest
python /share/nas1/yuj/script/chloroplast/advance/pi/cp_full_genome_pi_plot.py -i pi.stat.xls -o pi.full.png -yl 0.08 -yn 0.008 -xn 8300 -p 0.024 -lr xxx -ir xxx -sr xxx
# 需要输入边界,修改-p阈值,yl和yn手动输的不对会自动修正
-i [infile], --infile [infile]
infile
-o [outfile], --outfile [outfile]
outfile
-p [threshold], --pi_value_threshold [threshold]
pi大于阈值显示基因名
-yl [ylim], --y_lim [ylim]
Y轴最大值
-xn [x_tick_interval], --x_tick_interval [x_tick_interval]
X轴间隔
-yn [y_tick_interval], --y_tick_interval [y_tick_interval]
Y轴间隔
-lr [lsc_right], --lsc_right [lsc_right]
lsc右边界
-ir [irb_right], --irb_right [irb_right]
irb右边界
-sr [ssc_right], --ssc_right [ssc_right]
ssc右边界
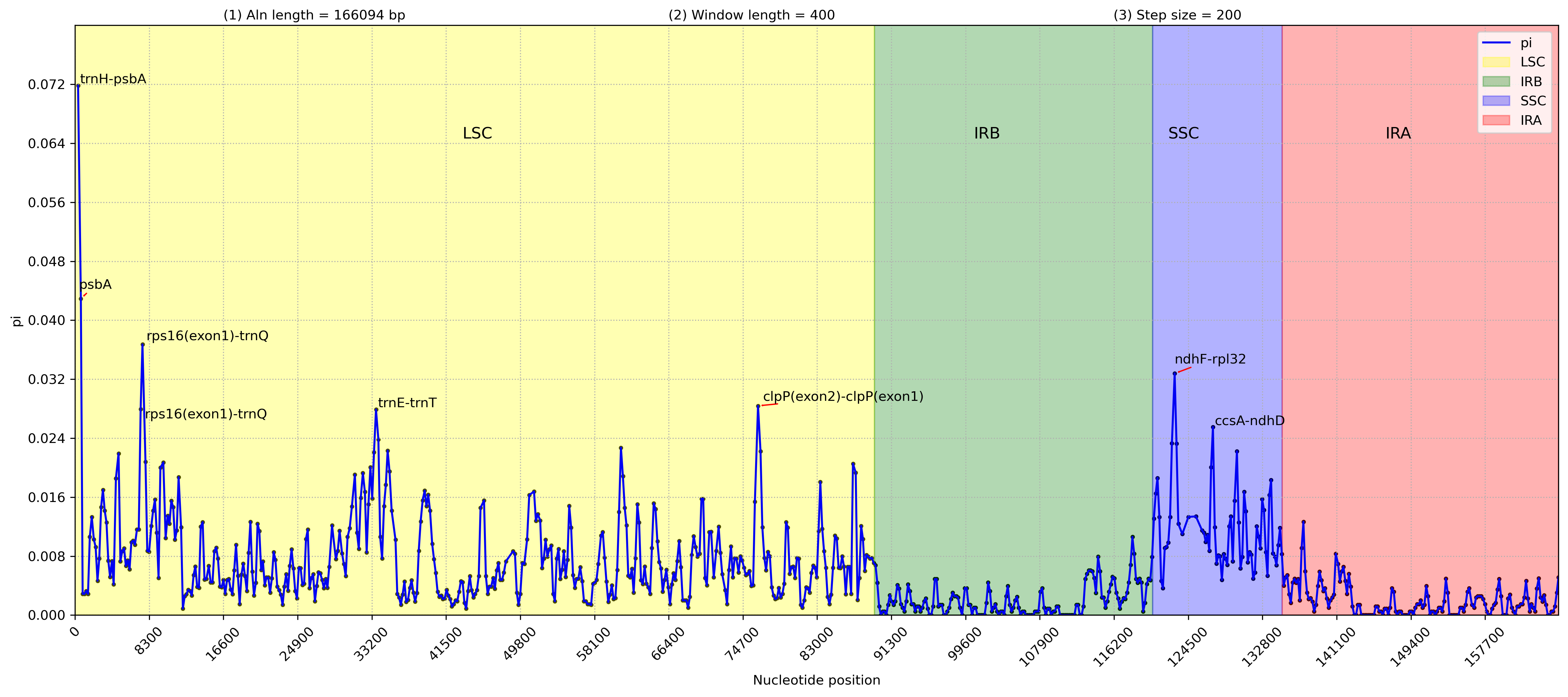
3.基因名(-i2 单独输入比对后的基因起止)
1.比对
mafft --auto --quiet --thread 30 allinone.fa > allinone.aln # 有点费时
perl /share/nas6/xul/program/mt2/phytree/gene_tree/src/fasta2line.pl -i allinone.aln -o all.fasta.line
2.滑窗计算
python3 /share/nas1/yuj/script/chloroplast/advance/pi/full_genome_calculate_pi.py all.fasta.line 600 50 > pi.stat.xls # 800 100看起来差不多
3.获取比对后基因位置
python3 /share/nas1/yuj/script/chloroplast/advance/pi/position_mapping.py -i all.fasta.line -p Ganoderma_sinense -i2 *.gbk.ann -o2 gene.range.info
4.绘图
mamba activate pylatest
python3 /share/nas1/yuj/script/chloroplast/advance/pi/cp_full_gene_pi_plot.py -i1 pi.stat.xls -i2 gene.range.info -o pi.gene.png
# 凑合用吧
3.4 边界分析
3.4.1 CPJSdraw(新版)
## 这一步的两个程序
perl /share/nas6/xul/program/chloroplast/irscope/CPJSdraw/script/create_input_file.pl ref_adv/gbk/*.gbk > irscope/gbk/cfg
perl /share/nas6/xul/program/chloroplast/irscope/CPJSdraw/bin/CPJSdraw.pl -i irscope/gbk/cfg -o irscope/irscope.svg
1.重新绘制
cd ../irscope
## 查找缺失基因
python3 /share/nas1/yuj/script/chloroplast/advance/cp_irscope.py -i ../../ref_adv/ -s ycf1 # 会显示ycf1的信息
perl /share/nas6/xul/program/chloroplast/irscope/CPJSdraw/bin/CPJSdraw.pl -i gbk/cfg -o irscope.svg
svg2xxx -t png -dpi 600 irscope.svg && svg2xxx -t pdf irscope.svg
2.修改图片里物种名字
FEATURES Location/Qualifiers
source 1..156901
/db_xref="taxon:2321412"
/mol_type="genomic DNA"
/organelle="plastid:chloroplast"
/organism="Camellia lienshanensis NC_067086.1" #### 改这里
# 一些对高级分析物种gbk的修改同3.4.2
3.4.2 Irscope(旧版)
原版
# 可能用到的其他脚本:
/share/nas6/xul/program/chloroplast/bin/cp_format_gbk.pl # 矫正gbk
/share/nas6/xul/program/chloroplast/bin/cp_gbk2fasta.pl # gbk生成fa
/share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/annotation/src/to_genbank.pl # 生成gbk
$ perl irscope_v3.1.pl -i cfg -p # 对应该步命令,-p假基因
# 1.画假基因图 2.查找所有基因
1.查看边界分析图片
gbk路径,修改的话,要改ref_adv里面的
图画了好几遍,都有问题,排除ref_adv里gbk问题后,一定要注意 cfg里填的ir区域位置,有可能是错的!!!!!!
2.修改cfg文件
2.1序列开头没有ir区的情况
改cfg里的分界,用ir程序的结果,一般情况可以100相似,但有的时候不是完全反向互补,即该物种ir区相似性不是百分百
gbk]$ ir AY916449.1.fasta
gbk]$ nucmer AY916449.1.fasta AY916449.1.fasta
gbk]$ show-coords out.delta # 获得正确的ir区位置
2.2如果ir区出现在开头,就需要矫正
------ fasta/MH909600.fasta ------
LSC:14-89921
IRb:89922-115951
SSC:115952-134528
IRa:134529-160545,1-13
perl /share/nas6/xul/program/chloroplast/bin/cp_format_gbk.pl -f .../NC_054249.1.gbk -s 14 # 矫正gbk -s要设定的正确起始位置
只修改一个文件时,就放在/gbk里不动,运行后生成/gbk/new_gbk/MW039136_no_IR.gbk
如果一堆要修改时,每个文件都新建一个目录放进去,使用时该命令 -i ./gbk
/share/nas6/xul/program/chloroplast/bin/cp_gbk2fasta.pl -i file -o outdir #gbk生成fa
/share/nas6/xul/program/chloroplast/bin/cp_gbk2fasta.pl -d gbk/ -o outdir
填入新的ir区位置
3.查找所有基因
$ perl /share/nas6/xul/program/chloroplast/bin/cp_Annotation.pl -i1 xxx.fasta -i2 .../ref/gbk/xxxxxx.1.gbk # 根据参考gbk查找当前fasta文件的注释
# 以ycf1为例
irscope/gbk]
$ cp_annotation_one_gene_by_ref_gbk2.pl -g ycf1 -i1 Quercus_hypargyrea_MW450871.1.fasta -i2 Quercus_hypargyrea_MW450871.1.gbk
$ vim ycf1
$ blastn -outfmt 6 -query ycf1 -subject Quercus_hypargyrea_MW450871.1.fasta
# editplus修改
# 真正起作用的是CDS那一栏,而假基因没有那一栏,只有gene一栏(旧版本)
# 新版本起作用的是gene一栏,因此rps19要更改
"""ycf1"""
gene 111629..112653
/gene="ycf1"
/pseudo
"""rps19(旧版本)"""
CDS 157301..157579
/gene="rps19"
"""rps19(新版本)"""
gene 136498..136779
/gene="rps19"
"""trnH-GUG"""
complement(join(1..59,157596..157611)) →→→ complement(157596..157611,1..59)
4.修改图片里物种名字(旧版)
LOCUS Hibiscus_syriacus_MH330684.11 161025 bp DNA circular PLN
DEFINITION Hibiscus syriacus MH330684.12 chloroplast, complete genome.
ACCESSION Hibiscus_syriacus_MH330684.13
VERSION .
KEYWORDS .
SOURCE chloroplast Hibiscus syriacus MH330684.14
ORGANISM Hibiscus syriacus MH330684.15 ########################### 修改此处
5.再次画图
6.根据结果调整显示顺序(可选) 修改cfg里的顺序
3.5 同源性比对
3.5.1 Mauve
pi]$ cat ../annotation/*/gene_anno/*.gbk ../../ref_adv/gbk/*.gbk > ../mauve/all.gbk && cd ../mauve
mauve]$ Mauve &
PS:不显示基因,优先考虑把组装的样品属名简写
1.ALL.GBK里的顺序为默认显示顺序
可以在软件里修改
也可以在文件夹里把gbk全部改成物种名,进行排序
2.运行程序出错的话,指 能打印日志的界面出错
重新cat合并
3.如果发现名字没有加载,太长了,去all.gbk删名字
LOCUS ACCESSION改这俩,而且一般是我们生成的
20220303补充:如果没有基因加载,也是物种+登录号名字太长了,改一下
通常是因为自己重新注释了,流程里用 物种+登录号 作为名字导致的
20220822补充:如果仅所有参考可加载基因,说明问题出在组装的gbk上
把组装的属名简写,一般就正常了
20221012补充:同上
4.图片正常显示,修改名字,查找如下字段
/organism=" "
FEATURES Location/Qualifiers
source 1..160819
/mol_type="genomic DNA"
/note="type: DNA"
/organelle="plastid:chloroplast"
/organism="Pecans5"##################################此处改名字
5.trna跨首尾,图里会出现一条绿色的线,对应位置去掉末尾或者开头部分
6.调整选项
LCB outlines■
Similarity plot■
Similarity ranges■
Solid LCB coloring
LCB strikethrough lines■
LCB connecting lines■
Chromosome/contig boundaries■
Show mouse highlighting
Draw attributes (histograms)■
7.导出图片路径(注意!!!不要选中框里的数值,会自动复制到剪切板)
/share/nas1/yuj/project/GP-20220413-4204_20220418/analysis/mauve/mauve
8.修图 长度显示不完全
3.5.2 mVISTA
1.网站
http://genome.lbl.gov/vista/index.shtml
2.转换注释
perl /share/nas1/yuj/script/chloroplast/advance/get_mVISTA_format_from_GenBank_annotation.pl -i gbk -p .gbk # -i输入文件夹 -p输入文件后缀
3.上传所有物种fasta + ref物种注释
把ref物种fasta上传两遍,因为第一个参考作参考后不显示该基因组
4.选择Shuffle-LAGAN比对
5.结果页面,选第一个物种,点击"VISTA-Point",查看一下
6.结果页面,点击第一个物种右侧pdf下载
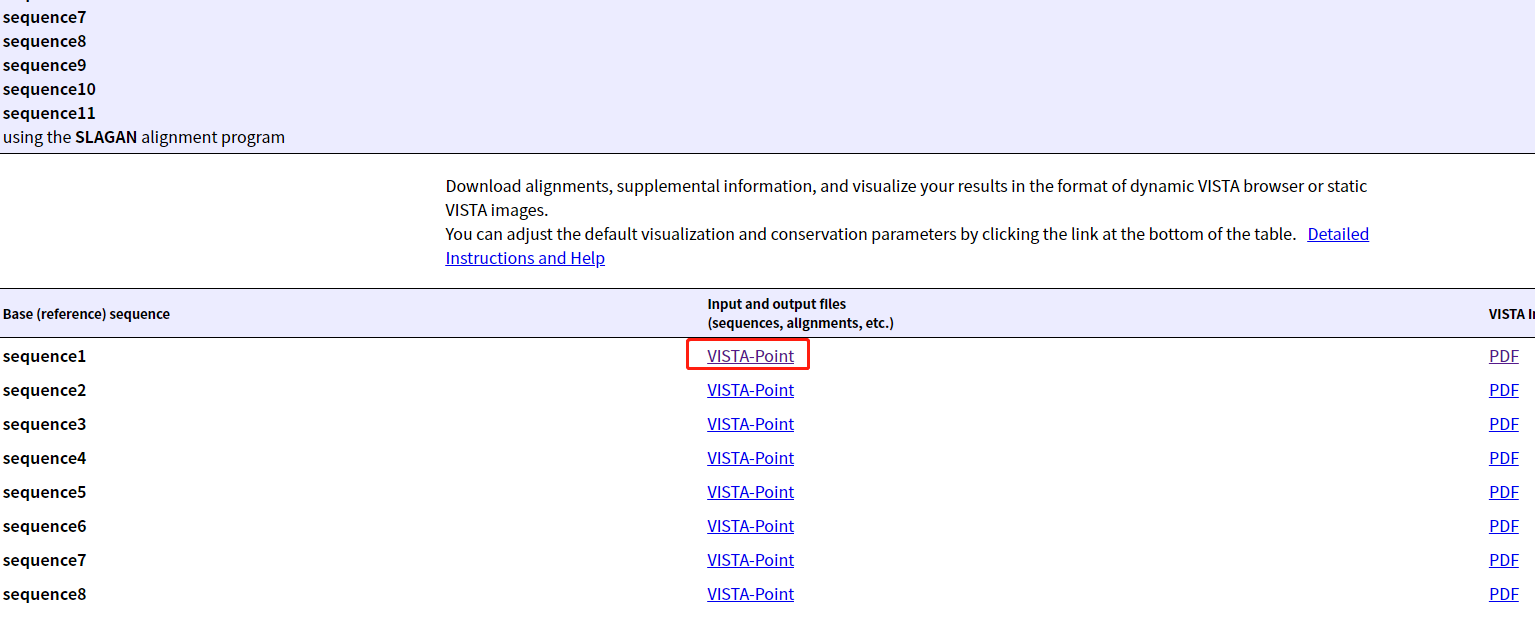
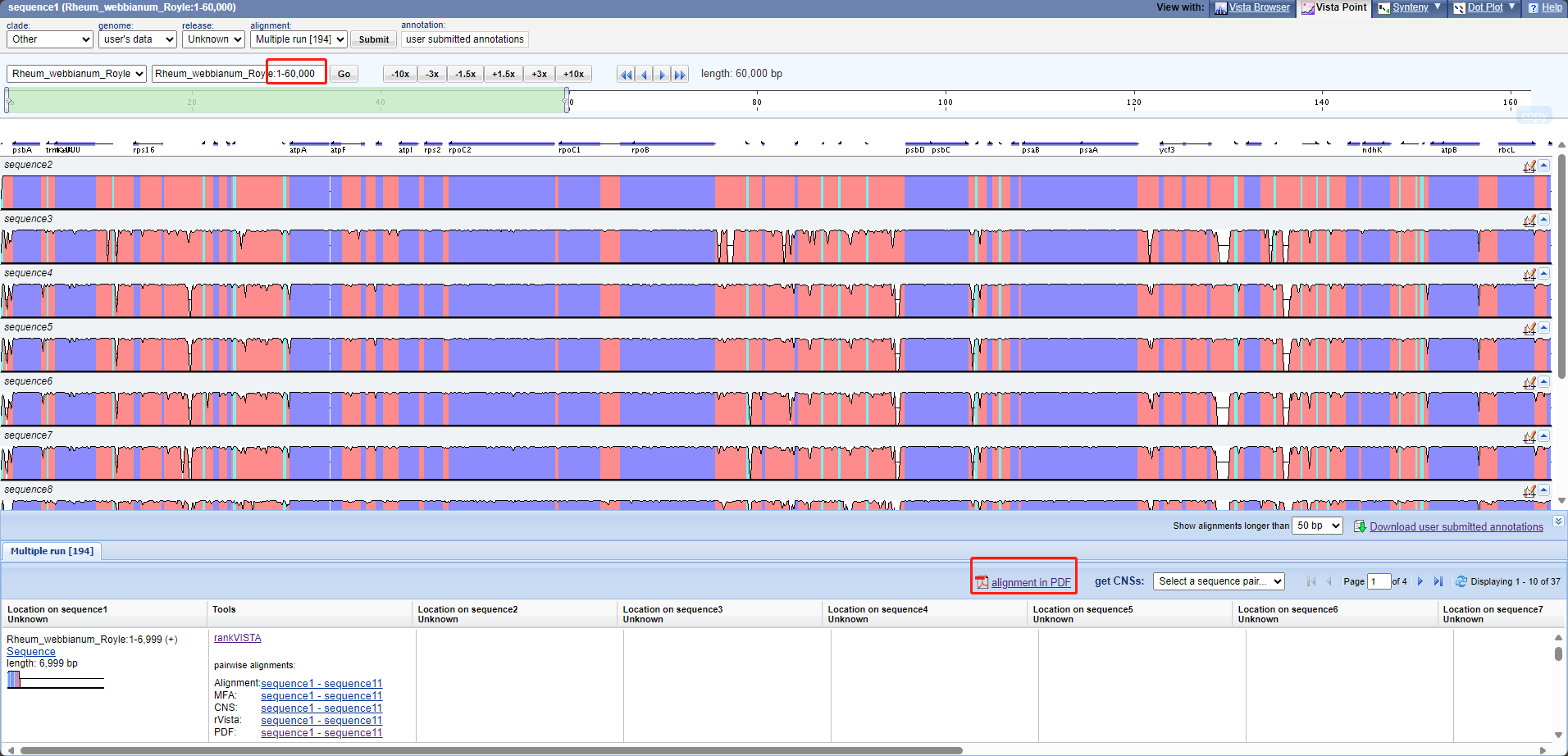
3.6全局比对(snp indel)
# 3354项目有全局比对(SNP indel)
analysis]$ cd global_align/
$ perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/script/global_align/global_align.pl -i 组装出的基因组(一般是第一个) -a 一般是第一个物种注释(没有也可以运行) -r 参考基因组/其他物种1.fa -o global_align
3.7 基本信息统计
1.genome size
for i in *.fasta;do fl $i;done
2.region length
for i in *.fasta;do perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/annotation/src/IR_SC_RegionFinder.pl -i $i -p ${i%.fasta} -o ./ & done
for i in *.region.fa;do echo "--------"$i; fl $i;done
3.GC content(%)
for i in *.fasta;do echo $i;ir.py -i $i -gc |tail -n 2;done
4.Number of genes
for i in *gbk;do st $i;done
四.整理结果
## 检查图片
mkdir fig && cp analysis/annotation/*/ogdraw/*.dpi300.circular.png analysis/annotation/*/asmqc_dir/coverage_plot/*.png analysis/annotation/*/asmqc_dir/cmp.dotplot.png fig
1.标准+高级
perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/cp_pip_dir.pl -i analysis -o complete_dir &
2.仅HIFI
perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/cp_pip_dir_hifi.pl -i analysis -o complete_dir
## 只有高级分析
perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/cp_pip_dir_only_advance.pl -i analysis/ -o adv_complete_dir
rm -rf complete_dir/03Asmqc/*/*.depth.svg
rm -rf complete_dir/03Asmqc/*/sample.coverage.svg
:set nu vim显示行数
五.生成报告
1.修改报告配置文件
cp /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/html_report/cp.report.cfg report.cfg
for i in `cat ann.cfg |grep gbk | cut -f 4`;do echo "#####################"$i && head $i;done
2.二代 标准+高级分析
perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/html_report/report2xml.yelvti.pl -id complete_dir -cfg report.cfg
3.二代 只有高级分析
perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/html_report/report2xml.yelvti_advance.pl -cfg report.cfg -id adv_complete_dir
4.仅HiFi数据
perl /share/nas6/pub/pipline/genome-assembly-seq/chloroplast-genome-seq/v1.2/html_report/report2xml.yelvti_hifi.pl -id complete_dir -cfg report.cfg
六.方法描述
## pi
1.方法
pi(核酸多样性)能揭示不同物种核酸序列的变异大小,变异度较高的区域可以为种群遗传学提供潜在的分子标记。
使用mafft软件(--auto模式)对不同物种的叶绿体基因组序列进行全局比对,
使用DnaSP v6.12.03(http://www.ub.edu/dnasp)计算多序列的核酸多样性pi值。
2.结果
├── 01allinone.fa是87的样品序列
├── 02allinone.aln是对齐后的序列
├── 03pi.stat.xls是计算的pi值
├── 04gap_pos_for_real_pos.xls是对齐后的位置(有gap)和原序列的位置对照,以Anoectochilus_papillosus为参考
├── 05pi.gene.png是绘制的图片
└── 05pi.gene_gene_avg_pi.xls是每个基因的平均pi值,根据03pi.stat.xls来的